
Pathways of Prevention: A Scoping Review of Dietary and Exercise Interventions for Neurocognition
Abstract
Alzheimer’s disease and related dementias (ADRD) represent an increasingly urgent public health concern, with an increasing number of baby boomers now at risk. Due to a lack of efficacious therapies among symptomatic older adults, an increasing emphasis has been placed on preventive measures that can curb or even prevent ADRD development among middle-aged adults. Lifestyle modification using aerobic exercise and dietary modification represents one of the primary treatment modalities used to mitigate ADRD risk, with an increasing number of trials demonstrating that exercise and dietary change, individually and together, improve neurocognitive performance among middle-aged and older adults. Despite several optimistic findings, examination of treatment changes across lifestyle interventions reveals a variable pattern of improvements, with large individual differences across trials. The present review attempts to synthesize available literature linking lifestyle modification to neurocognitive changes, outline putative mechanisms of treatment improvement, and discuss discrepant trial findings. In addition, previous mechanistic assumptions linking lifestyle to neurocognition are discussed, with a focus on potential solutions to improve our understanding of individual neurocognitive differences in response to lifestyle modification. Specific recommendations include integration of contemporary causal inference approaches for analyzing parallel mechanistic pathways and treatment-exposure interactions. Methodological recommendations include trial multiphase optimization strategy (MOST) design approaches that leverage individual differences for improved treatment outcomes.
SCOPE OF AGE-RELATED NEUROCOGNITIVE DECLINE
The number of older (aged > 65) adults in the United States is projected to double by 2030, representing an immense demographic shift that has been referred to as the ‘silver tsunami’[1]. Along with an increasingly older and complex general population, rates of age-associated neurocognitive impairment, stroke, and Alzheimer’s and Related Dementias (ADRD) are projected to increase in tandem, representing a looming source of public health expenditures [2]. ADRD is among the mostly burdensome and costly public health concerns in the United States. As treatments for coronary disease and cancer have increased life expectancies among aging adults, the risk of ADRD has continued to increase, with a projected increase in prevalence from 26.6 million to over 106 million worldwide by mid-century [3–5]. Moreover, many more individuals will develop cognitive impairment that substantially impairs quality of life without reaching criteria for dementia, including mild cognitive impairment (MCI) and cognitive impairment no dementia (CIND) [52]. Prior estimates have suggested that ADRD may account for up to $215 billion annually in public health expenditures, [6] with an anticipated increase in the coming decades. The public health cost of ADRD is particularly troublesome as the presence of ADRD is not only detrimental to afflicted individuals but often has dramatic ramifications for the quality and productivity of life among caregivers, many of whom have to substantially restrict or discontinue their participation in the workforce in order to provide care [7]. In addition, most older adults reportedly fear the development of ADRD more than cardiovascular disease (CVD) or other common chronic diseases, [8, 9] suggesting that strategies to mitigate ADRD risk have critical importance for both the quality and quantity of life for an exponentially increasing number of ‘baby boomers’ now at risk for ADRD [10–12].
The public health cost of ADRD is particularly troublesome as the presence of ADRD is not only detrimental to afflicted individuals but often has dramatic ramifications for the quality and productivity of life among caregivers, many of whom have to substantially restrict or discontinue their participation in the workforce in order to provide care [7]. In addition, most older adults reportedly fear the development of ADRD more than CVD or other common chronic diseases, [8, 9] suggesting that strategies to mitigate ADRD risk have critical importance for both the quality and quantity of life for an exponentially increasing number of ‘baby boomers’ now at risk for ADRD [10–12]. The increasing prevalence of ADRD is particularly concerning as pharmacological treatments to mitigate or prevent ADRD have been largely unsuccessful, with a 99% failure rate among novel pharmacotherapies [13]. Indeed, despite recent trials reporting tentatively positive findings, the consistent lack of efficacy observed from interventions among symptomatic older adults has led several large pharmacological entities to reallocate resources away from therapeutic pursuits due to a perception of futility [14–16]. Accordingly, a burgeoning body of evidence suggests that targeting modifiable risk factors in midlife may hold promise for mitigating or even preventing ADRD in later life [17–23]. For example, the Lancet Commission recently concluded that optimal treatment of modifiable risk factors holds the potential to delay or prevent up to one-third of ADRD cases [2, 24]. However, due to a relative paucity of randomized controlled trials, similar systematic reviews have yielded far less positive conclusions [25]. Beyond the potential merit of lifestyle change for preventing ADRD, optimizing modifiable risk factors is increasingly recognized for its importance in protecting from more normative decline in the context of cognitive aging, which has implications across all older adult populations [1, 26]. The present review will provide a narrative review of contemporary literature examining lifestyle characteristics and neurocognition, as well as innovative opportunities to improve mechanistic inferences from lifestyle trials attempting to mitigate ADRD risk and improve neurocognition.
LIFESTYLE, NEUROCOGNITION, AND ADRD: IMPORTANCE OF MODIFIABLE RISK FACTORS
A burgeoning body of evidence suggests that targeting modifiable risk factors in midlife may hold promise for mitigating or even preventing ADRD in later life [17–20]. For example, recent consensus statements by the NINDS, NIA, and American Alzheimer’s association have all advocated an increased focus on ADRD prevention, primarily focusing on three overarching risk factors: physical inactivity, ‘Western’ dietary patterns (e.g. high intake of saturated fat and complex carbohydrates, and low intake of fruits and vegetables), and poorly controlled cardiometabolic (CVD) risk factors. In contrast to some ADRD risk factors, such as age and genetic predisposition, these lifestyle-related dimensions all share an important characteristic: they are modifiable. CVD risk factors include an array of risk factors shown to increase risk of ADRD through vascular and/or metabolic risk, including components of metabolic syndrome (hypertension, obesity, diabetes or insulin resistance, hyperlipidemia, elevated triglycerides), smoking, atrial fibrillation, and left ventricular hypertrophy. In the United States, common CVD risk factors (e.g. hypertension and obesity) affect nearly 2/3 of adults and are independently predictive of cognitive decline and ADRD [10]. Individuals with elevated CVD risk factors in midlife are 2-3 times more likely to develop dementia, [17, 18, 27, 28]. leading many investigators to characterize CVD components as primary targets for dementia prevention initiatives [11]. Because AD and dementia are projected to constitute the largest public health burdens in the U.S. within two decades and pharmacological treatments have proven ineffective, the ability to curb cognitive deterioration carries tremendous public health importance [2, 29]. Indeed, although estimates related to potential prevention of ADRD are inherently imprecise, reductions in modifiable risk factors by 10–25% could reduce up to half of AD cases worldwide, representing millions of individual cases [30]. An integrated understanding of the effects of diet and exercise on neurocognitive functioning is therefore critical to guide prevention efforts over the coming decades.
BEHAVIOR AND THE BRAIN: CONCEPTUAL FRAMEWORKS LINKING LIFESTYLE AND NEUROBEHAVIORAL FUNCTIONS
“A system is a set of things— people, cells, molecules, or whatever— interconnected in such a way that they produce their own pattern of behavior over time. The system may be buffeted, constricted, triggered, or driven by outside forces. But the system’s response to these forces is characteristic of itself, and that response is seldom simple in the real world.”[31]
Neurobehavioral Considerations
Over the past two decades, an explosion of empirical evidence has been published linking lifestyle behaviors to brain function [19, 32–36]. Indeed, great strides have been made at multiple levels of inference, including randomized trials of clinical populations, [37–41]. mechanistic studies within preclinical older adults using neuroimaging modalities, [42–44] and animal studies linking neurobehavioral changes to alterations in mitochondrial structure, [45–47] neurotrophic, [48, 49] and metabolic function [50, 51] to underlying changes of neuropathological pathways [52–54]. While exciting, the abundance of data has made a systematic synthesis of the linkages between diet, exercise, and neurocognition more difficult. One specific difficulty has been delineating overlapping mechanistic pathways by which change in lifestyle may alter the brain and, as a behavioral corollary, neurocognitive function. Moreover, prevailing biomarker models of preclinical ADRD progression increasingly suggest that subtle, systemic ADRD changes occur over decades, with alterations in neurocognitive function only observable in the final stages of the process [16, 55, 56].
Translational studies across animal and human models are potentially compounded by inherent and well-characterized neurobiological differences across animal and human studies, in which the behavioral markers most sensitive to the effects of aging [57–59] and CVD risk factors [10, 60] in humans are also the most distinct and hardest to measure in animals (e.g. higher-order executive functions). For example, while the most anterior brain regions in humans (e.g. orbitofrontal cortex) and associated projections have widely studied neuropsychiatric relevance, [61–65] these brain regions have less representation in rodents, making translational behavioral inferences more difficult [66]. Therefore, while critically informative for elucidating mechanistic underpinnings, animal studies have necessarily prioritized behavioral markers of learning and memory, [67] which are critically reliant on mesial temporal lobe structures in humans.
In humans, episodic memory performance is closely tied to integrity within mesial temporal lobe structures, providing the earliest and most prototypical impairment observed in preclinical AD, often with associated cortical atrophy and ventricular enlargement [68, 69]. In contrast, the most common age-related neurocognitive changes occur on tests of complex processing speed, [57, 70] which are subserved by a small array of frontal-subcortical circuits (FSCs), some of which are preferentially vulnerable to microvascular and ischemic damage [71]. Underscoring their complexity, the FSCs all have direct and indirect pathways across the prefrontal cortex, corticocortical connections with other circuits, and more distal connections with outside brain regions [72]. While interconnected, FSCs carry various degrees of importance in facilitating distinct components of ‘executive functions’ depending on the specific needs of the task. Anterior injuries to the superior / medial FSC may experience impairments in task initiation, whereas damage to the dorsolateral prefrontal cortex are more commonly associated with impairments in set-shifting and working memory. FSCs also facilitate a delicate balance of neurotransmitter functions across various ‘reward system’ brain regions in the ventral striatum and caudate, which are particularly important in their sensitivity to various monoamines (e.g. dopamine). This may explain why neurocognitive improvements following exercise appear to preferentially improve executive functions and speed across diverse clinical populations, such as vascular cognitive impairment, [73]. Parkinson’s Disease (PD), [74] and Attention Deficit Hyperactivity Disorder (ADHD) [75]. While neurocognitive deficits across these disorders are etiologically distinct, they have shared dysfunction within neurocircuitry in the FSCs and particularly the ventral striatum and associated prefrontal cortex projections, which are critical to facilitating these complex neurocognitive functions.
Conceptual models
Several widely used conceptual models are useful in elucidating the complex associations between lifestyle factors and neurocognition. One widely used and conceptual framework is the scaffolding theory of aging (STAC) and its revision (STAC-R) [76–78]. This framework explicitly grapples with several important elements of the dynamic systems by which peripheral systems, structural markers of neuropathology, and functional neural pathways all act in concert facilitate neurocognitive function. It is worth remembering that, although an increasing focus is been placed on the identification of preclinical biomarkers the AD literature, the end goal of treatment remains the same: to preserve function through preserved neurocognition [79–81]. Importantly, many investigators hypothesize that the beneficial effects of lifestyle modification on neuroplasticity are occur primarily by strengthening compensatory functions of the brain, with more modest effects occurring through direct enhancement to neuropathological aspects of brain structure (e.g. cortical atrophy and white matter disease) [77]. However, the ability to recruit and retain new neurons is also dependent on the underlying health of the brain and supportive systems, such that there may be a ‘tipping point’ beyond which these compensatory changes are eclipsed by larger neurodegenerative changes.
An important corollary of this theory is that the impact of lifestyle on brain function may act through different mechanisms with a different time course is depending on the person’s age, neuropathological burden, and systemic health. For example, optimally managing cerebrovascular risk factors over the course of decades through exercise and diet would likely help sustain brain function by sustaining microvascular function [82]. In contrast, reducing cerebrovascular risk over the course of months among an older individual with significant cortical volume loss may not be helpful and could paradoxically worsen function if compensatory cerebrovascular remodeling has occurred within the neurovascular unit following decades of hypertension [83–85]. In essence, this theory allows for a dynamic, systems level approach to cognitive functioning that takes into account multiple factors often overlooked and more simplistic models.
Another important conceptual approach is the potential ability of lifestyle to strengthen neurocognition through its impact on reserve capacity, which has conventionally been conceptualized as cognitive reserve [86] and more recently extended to include metabolic [87–89] and brain reserve [90]. Reserve capacity reflects wide individual differences in the ability to withstand great amounts of neuropathological damage without apparent neurocognitive impairment, which may be reflected across multiple systems that all indirectly impact ultimate neurocognitive function. For example, markers of fluid intelligence, [91] greater efficiency of metabolic ‘cross-talk’ between the peripheral and central nervous systems (CNS), [92] and greater white matter integrity [93] all could be conceptualized as representing reserve capacities, measured across cognitive, metabolic, and structural brain systems. Although there appears to be wide agreement that reserve capacity is a critical component underlying the associations between diet, exercise, and neurocognition, [32, 89, 94, 95] there remains wide disagreement about how these concepts are defined and how they should be measured [80, 81, 96]. Nevertheless, it appears likely that lifestyle has a beneficial effect on the brain through its parallel impacts on brain structure, reserve capacity, and directly on neurotransmitter systems, which indirectly influence each other (Fig. 1).
Related conceptual frameworks with smaller empirical bases in humans include the effects of lifestyle on 1) hormesis and 2) compensatory repair functions within the brain facilitated by preserved lipid metabolism. Hormesis reflects the adaptive response of the brain to intermittent, low-dose environmental exposures (e.g. dietary energy restriction) that enhance systemic functional response capabilities [97, 98]. More recent evidence suggests that alterations in phospholipid metabolism may be particularly important to the progression of subclinical ADRD biomarkers of cortical atrophy [99]. CNS lipids have a critical, structural role in supporting neuronal integrity and it has been hypothesized that intact phospholipid metabolism may serve a critical compensatory mechanism underlying neuronal repair in preclinical adults. Alterations in phospholipid metabolism are prognostic of incident cognitive decline, conversion from normal cognition to mild cognitive impairment (MCI), and conversion from MCI to ADRD [99, 100]. Altered lipid metabolism is also potentially consistent with the increased rates of ADRD among individuals with different apolipoprotein (APOE) genotypes, as the APOE gene was originally discovered because of its lipid metabolizing properties. It is therefore possible that blunted peripheral metabolic function may impair the brain’s ability to offset normative neurodegeneration, with marked dysregulation of phospholipid metabolism occurring in the years immediately preceding clinical neurocognitive impairment [101–105].
Fig.1
Directed acyclic graph linking lifestyle modification to neurocognitive outcomes. As shown, lifestyle modification likely has beneficial effects on structural markers of neuropathology, reserve capacities, and more directly on neurotransmitter systems, all of which contribute to neurocognitive function as observed through behavioral testing. The associations between markers of reserve capacity and/or scaffolding (i.e. blood brain barrier integrity, cerebrovascular reactivity, neurogenesis) likely impact neurocognition indirectly through their influence on structural markers and by potentiating or blunting neurotransmitter systems.
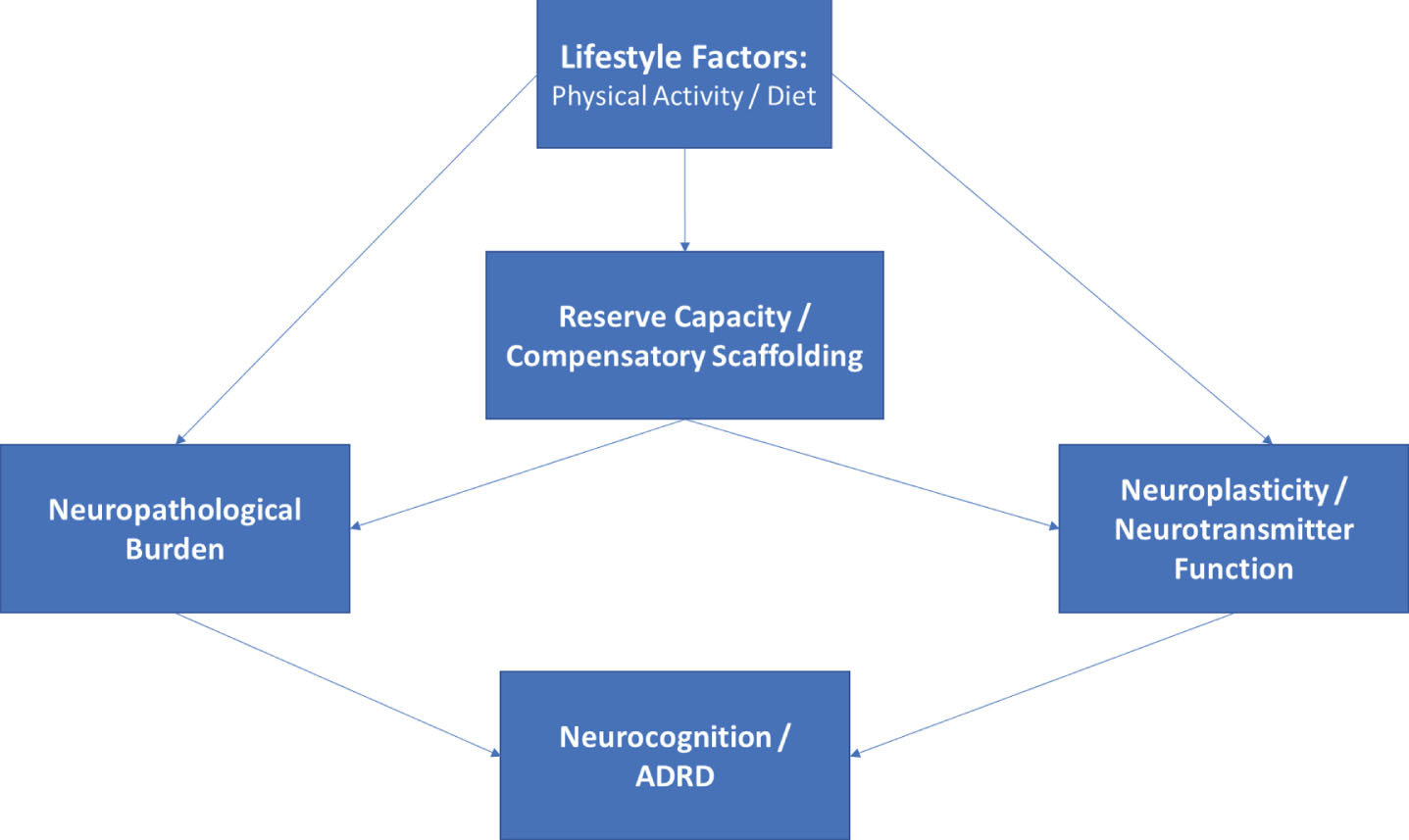
Conceptual frameworks have proven useful in guiding lifestyle-related intervention approaches, but have been used sparingly in generating falsifiable hypotheses. As discussed later in this review, this may owe partly to the inherent ‘systems-level’ nature of models following from models such as STAC-R, which posit that an individual’s level of neurocognitive function represents the brain’s ability to compensate for underlying disease burden. Implicit within this standard definition of brain reserve / resilience is a multi-level system in which function (neurocognition) is a product of disease exposure (e.g. advancing age, vascular risk), neuropathological burden (e.g. cortical atrophy), and cerebral compensatory factors (e.g. neurogenesis, synaptic plasticity). In other words, exogenous disease exposure causes both structural and functional degradation in the CNS, and neurocognition therefore reflects the degree of successful compensation exerted by an individual to sustain function in the presence of this underlying burden. Neurocognitive performance therefore reflects a gradient: it reflects both the amount brain function / reserve capacity exerted and the amount of underlying neuropathological / structural burden.
Dynamic conceptual models suggest several important and testable hypotheses. First, the association between markers of exposure and neurocognition should be mediated through their impact on underlying brain structure and compensatory function. For example, the association between advancing age and memory decline would be explained by alterations in structural (e.g. cortical atrophy and microvascular burden) and compensatory / functional (e.g. CNS metabolic function). In other words, an individual with greater vascular risk who (miraculously) does not evidence microvascular ischemic damage or impaired cerebrovascular reserve function would be hypothesized to have better neurocognition compared to an individual without evidence of these CNS sequelae, even if they were comparable in their degree of (peripheral) vascular risk. Similarly, greater white matter damage / leukoencephalopathy caused by factors unrelated to chronic vascular risk factors (e.g. CADASIL or multiple sclerosis) would be hypothesized to impair neurocognition even in the absence of vascular risk, because the primary mechanism of impairment (brain structure) is still impacted. To use a more concrete analogy, knowing that a car cannot get from point A to point B tells you little about whether the car’s impairments are structural (e.g. the frame is compromised) or functional (e.g. flat tires and out of gas). Designing intervention to make the car function would therefore be highly dependent on the type of impairment: filling up the gas is unlikely to be helpful if your car’s frame is compromised. As a corollary, lifestyle interventions designed to modify exposure variables (e.g. blood pressure) or that select participants based on their level of neurocognition may therefore overlook important individual differences in biomarkers of disease burden that could plausibly impact participants’ ability to improve brain function with treatment.
Another important and testable extension of these conceptual models is that structural markers of reserve should function as a constraining factor to any treatment-related improvements in neurocognition. Because structural reserve markers (e.g. atrophy) are less changeable and in some cases represent a marker of optimal functioning (e.g. educational status or premorbid intelligence), they may function to moderate the degree to which improving compensatory factors improves neurocognition. In other words, structural markers may impact the capacity of improvement, whereas improvements in functional markers of reserve may track more closely with neurocognitive changes. In some conceptual models, like STAC-R, lifestyle modification is thought to impact neurocognition primarily through its impact on compensatory factors, with smaller gains over time in structural neuropathological markers. As a corollary, improvements in neurocognition may be mitigated in the presence of greater neuropathological burden. Nevertheless, few trials attempt to collect or integrate individual difference in structural markers of disease burden into their inclusion criteria or account for them in their analytic plans [106].
PHYSICAL ACTIVITY, DIETARY PATTERNS, AND NEUROCOGNITIVE OUTCOMES
Lifestyle factors, such as physical activity and healthier dietary patterns, are increasingly recognized as potential contributors to ADRD risk [18, 107–111]. For example, meta-analytic studies of middle-aged adults have demonstrated that greater engagement in physical activity associates with significant reductions in ADRD risk, with individuals who exercise regularly demonstrating an approximate 40% reduced risk [112]. Prospective studies have also linked various dietary patterns to differential ADRD outcomes [110, 113, 114]. Multiple intervention studies have reported similar findings, with numerous meta-analytic reviews suggesting that individuals randomized to participate in aerobic exercise training experience modest but appreciable increases in cognitive performance [115–118]. These findings are reviewed briefly below.
Physical activity
Physical activity, including leisure time exercise, has gained increasing attention as a possible modifiable factor mitigating the risk of Alzheimer’s disease and cognitive impairment in later life [112, 119, 120]. Numerous epidemiological studies now suggest that greater levels of physical activity confer a lower risk of a dementia in later life, with even modest improvements and habitual activity demonstrating significant reductions in long-term risk. For example, in a meta-analysis of 15 prospective studies incorporating nearly 34,000 individuals, higher physical activity levels associated with a 38% lower risk of neurocognitive decline, with even low-to-moderate exercise levels conferring lower risk (35%) [112]. Discrepant findings have also been reported, however, with a recent study from the Whitehall II study failing to find a protective association between physical activity and risk of ADRD nearly 30 years later [121]. These discrepant findings may be influenced, in part, by differing risks between ADRD subtypes, with AD and vascular dementia (VaD) differing to various degrees in their underlying pathophysiologic risk profiles. For example, a more recent meta-analysis of prospective studies demonstrated that physical activity was protective against all-cause dementia (OR = 0.79 [0.69, 0.88]) and cognitive decline (OR = 0.67 [0.55, 0.78]), with the greatest benefits for development of AD (OR = 0.62 [0.49, 0.75]), whereas risk of VaD was not associated with physical activity (OR = 0.92 [0.62, 1.30]) [122]. An interesting but inconsistent finding within published exercise trials is the potentially differential effect based on genetic risk for AD (APOE) [123, 124]. Although results have been mixed, several studies have demonstrated that the beneficial effects of aerobic training may be greater among individuals with greater AD genetic risk, [123] although others have failed to replicate these subgroup differences [125, 126]. Taken together, these findings suggest that being physically active in middle-age is likely protective against future ADRD, although there is wide individual variation across individuals that remains to be understood, as discussed in later sections below.
Physical activity interventions also have tended to report beneficial effects, [41, 117, 127–129]. with some notable exceptions [36]. Results have generally suggested that improvements in neurocognition following aerobic training are strongest in frontal-subcortical functions, including attention and executive functions, although improvements across other domains also have been noted [117, 118]. Similar findings have been reported from resistance training interventions, with participants randomized to exercise demonstrating improvements relative to controls [130] and a tendency for the pattern of improvements to favor frontal-subcortical functions [131, 132]. As discussed in more detail below, the largest source of variation across studies appears not to be whether they can improve neurocognition, but whether the observed improvements can be attributed to changes in fitness or other downstream mechanisms. For example, multiple studies examining the physical activity and neurocognition association have failed to demonstrate dose-response benefits [112, 117, 133–135] or an association between improved aerobic fitness (the putative treatment mediator for aerobic training) [36, 136]. The comparable benefits observed from resistance training may therefore suggest that downstream changes (e.g. metabolic function or neurotrophins) represent a more plausible mechanism underlying the observed treatment benefits.
Dietary patterns
An extensive body of evidence, including hundreds of trials, has attempted to improve neurocognition through dietary supplementation. As reviewed in detail elsewhere, areas of intervention tended to focus on antioxidants (vitamins A, C, and E), B vitamins and folate, and polyunsaturated fatty acids (e.g. omega-3 s). Although the details of this large body of evidence are beyond the scope of the present paper, systematic reviews of supplementation trials have generally produced equivocal findings with one exception: individuals who are deficient in a various nutrient (e.g. vitamin B) benefit from supplementation to normalize their levels. Beyond this, supplementation does not appear to improve neurocognition and may even have adverse effects in some individuals. An emerging body of evidence has shifted focus form specific nutrients to overall dietary patterns, with a focus on the Mediterranean diet (MeDi), the Dietary Approaches to Stop Hypertension (DASH) diet, the Mediterranean-DASH for Neurodegenerative Delay (MIND) diet, and caloric restriction.
Dietary patterns have also gained interest, as a growing number of prospective studies have demonstrated that better adherence to dietary patterns with higher intake of fruits, vegetables, whole grains, and reduced intake of saturated fat and complex carbohydrates appear to be associated with reduced ADRD risk [137–141]. For example, greater adherence to the DASH diet, [142–147] MeDi, and MIND [148–150] have all been associated with lower ADRD risk. However, there have also been null findings, with recent systematic reviews reporting that evidence linking dietary patterns to ADRD outcomes remains inconclusive [113, 151, 152]. Importantly, although the MeDi and DASH were originally characterized for their beneficial impact on CVD risk factors and CVD outcomes, few studies have provided compelling evidence that the observed associations between dietary patterns and ADRD are mediated through vascular pathways, [148, 153] with multiple, indirect mechanisms connecting dietary behaviors to cognitive outcomes [144, 154].
Mediterranean diet
One of the most widely studied dietary patterns is the MeDi. The MeDi was originally identified for its characteristic pattern among individuals living in European countries surrounding the Mediterranean sea, who also consistently exhibit a low incidence of cardiovascular disease. Accordingly, the MeDi is typified by greater levels of fish intake, fresh fruit and vegetables, unsaturated fatty acids, and modest but regular consumption of wine [153, 155]. The majority of prospective cohort studies have found a protective association between the MeDi and neurocognitive outcomes [137, 156, 157]. For example, a prior meta-analysis combining results across five studies found that higher adherence to the MeDi diet (highest tertile) was associated with a 27% reduced risk of MCI and a 36% reduced risk of AD among cognitively normal adults [158]. The majority of existing observational evidence suggests that the beneficial effects of MeDi on brain outcomes are mediated by reduced cerebrovascular risk and incidence of brain infarcts on neuroimaging assessment [159]. As detailed below, results from the PREDIMED trial, [160, 161] are one of the only RCTs available to assess the benefits of MeDi diet in an intervention context, though several ongoing trials are currently underway. In addition to available trial data, a recent meta-analysis of prospective cohort studies suggested that the association between MeDi dietary pattenrs and ADRD exhibits dose-response characteristics, suggesting that increasing levels of adherence may have additionally protective effects [162].
Dietary approaches to stop hypertension (DASH)
Similar to the MeDi, the DASH diet was originally designed for its beneficial effects on blood pressure and associated hypertensive outcomes. The DASH diet emphasizes consumption of fresh fruits and vegetables, whole grains, low-fat dairy products, modest meat consumption, and modest alcohol consumption. In contrast to the MeDi, the DASH places greater emphasis on reducing dietary salt intake and does not incorporate regular alcohol consumption. Greater DASH adherence has also been associated with greater blood pressure reductions [163] and reduced risk of stroke, [164] underscoring its potential benefits to cerebrovascular mechanisms of neurocognitive impairment. Preliminary evidence suggests that individuals who are more adherent to a DASH-style diet have a lower incidence of cognitive decline [144, 146]. For example, DASH diet adherence was also associated with lower rates of cognitive decline among older adults participating in the Memory and Aging Project during a 4-year follow-up [146, 165].
Mediterranean-DASH (MIND) Diet
An emerging dietary pattern of interest is the MIND diet, which combines aspects of the MeDi and DASH diet with an emphasis on dietary components linked to neuroprotection and dementia prevention [166–168]. For example, the MIND diet has a more explicit emphasis on dietary components with polyphenolic effects, such as blueberries, red wine, and dark chocolate [169]. The MIND diet has been associated with a lower incidence of neurocognitive decline [150, 168, 170] and ADRD [169–172]. Indeed, recent evidence suggests that the MIND dietary pattern is associated with more protective effects when compared to either the MeDi or DASH directly [165–167]. Although no clinical trials have focused on modifying dietary patterns using MIND specifically, investigators are increasingly integrating this pattern of dietary intake into preventive efforts aimed at lowering the incidence of ADRD through dietary modification [173].
Caloric restriction / intermittent fasting
Lower caloric intake is historically one of the most widely studied aspects of dietary intake believed to improve brain outcomes. While early studies were almost exclusively in animal models, an accumulating body of data suggests that lower caloric intake may be associated with lower risk of cognitive decline, [174, 175] and that intentional weight loss through caloric restriction may improve neurocognition [176]. For example, both cross-sectional [177] and prospective studies have suggested that caloric intake is associated with ADRD risk, [178] and that this association may vary based on genetic risk for AD [179]. In addition, several RCTs have examined the impact of caloric restriction on neurocognition among humans, with mixed results [178, 180–182]. In one study, a three-month caloric restriction intervention improved memory performance among fifty healthy, elderly adults who were either normal weight or overweight [178]. Following three months of treatment, individuals randomized to the caloric restriction group showed improvements in verbal memory performance. In addition, these improvements were associated with decreased levels of fasting insulin and C-reactive protein and were strongest among participants with the best adherence. Similar findings were noted in recent post hoc analyses from the CALERIE trial, [183] in which improvements on some neurocognitive subtests were noted among individuals in the caloric restriction among obese adults [184]. Moreover, improvements in neurocognition were associated with peripheral inflammation levels, [185] suggesting a possible mechanism linking caloric restriction and neurocognitive improvements. Despite these positive findings, three other trials examining caloric restriction failed to find significant effects, which may due to the younger age of participants in these trials [180–182].
In one of the more positive caloric restriction trials, Horie and colleagues demonstrated that a one-year caloric restriction trial in 80 obese adults with MCI improved cognitive function, with improvements observed across neurocognitive domains [186]. Improvements in neurocognition varied somewhat by age and APOE genotype, with participants aged 60–70 demonstrating the largest improvements in memory and verbal fluency, and APOE-4 carriers demonstrating the largest improvements in executive function. Most notably, improvements in neurocognition were associated with improved metabolic function (increased insulin sensitivity and leptin), reduced inflammation (C-reactive protein), and reduced energy intake (carbohydrates and fats). Similarly positive findings were reported from a small trial examining changes in brain structure and function among 37 obese, postmenopausal women randomized to an intensive 12-week low-caloric diet or a control group [187]. In a per protocol analysis, the authors excluded individuals in the intervention group who did not lose at least 10% body weight, as well as participants in the control group who lost more than 5% of body weight. Participants in the intervention group exhibited improved memory performance paralleled by increased gray matter volume in the inferior frontal gyrus and hippocampus, as well as augmented hippocampal resting-state functional connectivity to parietal areas. The effects appeared to be specific to transient negative energy balance, and were not detected after subsequent weight maintenance.
Other dietary patterns
Two closely related dietary patterns, with a much small literature base, include intermittent fasting and ketogenic dietary patterns. Intermittent fasting has been suggested to have beneficial effects on the brain by improving metabolic function through intentional manipulation of energy balance, as well as reducing inflammation and oxidative stress [188–190]. Intermittent fasting appears to produce comparable weight loss benefits when compared to traditional caloric restriction, [191] reduces cardiovascular risk factors, [192, 193] and has been associated with reduced risk of age-related neurocognitive deficits [194]. An additional benefit is that it may be easier for many individuals to comply with It also may be easier for many individuals to comply with, [195] increasing the likelihood of sustained adherence. Finally, the ketogenic diet (KD) has been examined, originally as a potential non-pharmacological strategy to reduce the frequency and severity of seizure activity in epileptic patients [196]. Through hermetic mechanisms, KD has been postulated to improve central metabolic function, [169, 197, 198] a critical factor in age-dependent neurocognitive decline [197]. Although very few studies have examined KD outside of epileptic samples, [199–201] mechanistic animal studies suggest that KD may improve glucose transport, with differential effects on the prefrontal cortex and hippocampus [202].
Randomized Trials Combining Exercise and Dietary Modification
Although individual lifestyle components appear to confer lower ADRD risk, preliminary evidence suggests additive benefits associated with each component, such that individuals engaging in regular exercise and healthier diets show the lowest risk [203–205]. Data from the WHICAP project demonstrated that participants engaging in physical activity or the MeDi diet had lower risk of Alzheimer’s Disease (AD), and participants engaged in both had the lowest prospective risk of AD [206]. Similar mechanistic findings have been demonstrated in animal models as well, with exercise facilitating the beneficial effects of some dietary components on brain outcomes [98, 207–212]. Preliminary data from multicomponent trials have reported similar findings, with both exercise and diet combining to improve neurocognitive performance [145, 213]. Similarly, in the recently completed Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) trial, [214] individuals randomized to the multicomponent intervention of exercise, diet, cognitive training, and monitoring of CVD risk factors experienced greater cognitive improvements. In contrast, recent data from the LOOK-AHEAD trial suggest that behavioral weight loss does not necessarily improve neurocognition [215–219] and may even have deleterious effects in older individuals, [220] despite improvements in some neuroimaging biomarkers of ADRD [221, 222]. Multicomponent trials are presented in detail below:
FINGER: The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) trial was one of the more positive RCTs to show improvements in neurocognition following lifestyle modification [214]. In this RCT, 1260 number of older adults (60–77) with cardiovascular risk factors were randomized to a two-year, multidomain intervention consisting of aerobic exercise training, dietary modification, cognitive training, and vascular risk monitoring. The control group received general health advice. Participants were 60–77 years of age, had a CAIDE (Cardiovascular Risk Factors, Aging, and Dementia) Risk Score of > 6 points, and had to exhibit some evidence of neurocognitive weakness based on screening using the CERAD neuropsychological test battery. The dietary intervention was conducted by study nutritionists and personalized across participants. Although the dietary intervention was developed to accord with Finnish dietary recommendations, the overall dietary pattern was similar to that proposed across other DASH and MeDi trials in that it consisted of high fruit and vegetable consumption, whole grains, low-fat daily products, reduced intake of refined carbohydrates, and consuming at least two portions of fish per week [223]. Weight reduction was not uniformly recommended, but a 5–10% overall energy reduction was implemented as necessary and the intervention group demonstrated overall weight reductions compared to controls [214]. In addition, participants engaged in a physical activity intervention, which was supervised for the first 6-months, and a 10-session cognitive training program facilitated by a study psychologist with additional computerized training provided to participants. The intervention was therefore uniquely comprehensive in its targeting of physical activity, whole-diet patterns, vascular risk reduction, and cognitive training.
The physical activity intervention consisted of both aerobic and resistance training and was modified from the Dose-Responses to Exercise Training (DR’s EXTRA), which used a personalized training target for aerobic exercise of 55–65% of peak oxygen consumption. Aerobic training was gradually titrated to increase duration and intensity over the course of the trial, beginning with 30–45 mins twice per week and incrementally increasing to exercise bouts of 45–60 mins 3–5 times per week [223]. Progressive muscle training was also individually tailored in terms of intensity and titration, but standardized its approach by using exercises for eight main muscle groups (e.g. knee extension and flexion, abdomen and back, rotation, upper back and arm muscles, and leg press for lower extremity muscles), as well as postural balance exercises.
Following two years of treatment, a modified ITT analysis revealed that the intervention group demonstrated improvements in physical activity, dietary composition, and reduced weight. Most importantly, there was a small but significant improvement in a composite marker of neurocognition, as indexed by a comprehensive neuropsychological test battery z-score. Closer examination revealed that the largest improvements were observed in processing speed and executive function, without consistent improvements in memory performance [213]. While statistically significant, it should be noted that the findings were modest as a standardized effect (ES = 0.13). In addition to the primary findings from FINGER, structural MRI markers were obtained at baseline, 1-year, and 2-years after randomization, providing an opportunity to examine intervention effects on established biomarkers of ADRD risk [224]. Surprisingly, no intervention effects were observed on changes in MRI markers. However, integration of MRI and cognitive data revealed that treatment-related improvements in processing speed were more pronounced among individuals with higher baseline cortical thickness and volumetric characteristics of mesial temporal structures with critical importance for AD risk, suggesting that the beneficial effects of treatment were strongest among individuals with less advanced neuropathological changes. These findings may suggest that lifestyle modification improves neurocognition, but must be initiated earlier in the disease process to yield appreciable benefits.
Notably, mechanistic data from FINGER demonstrated similar findings to some other trials below, with cardiorespiratory fitness associating most strongly with observed neurocognitive improvements in processing speed and executive function, but not memory [225]. Improved dietary patterns also predicted improvements in executive function, [226] whereas baseline dietary patterns (not change) associated with global cognitive improvements over the course of the intervention. Perhaps most notably, changes in neurocognition were not associated with APOE genotype [126] and greater CVD risk, although associated with volumetric characteristics of ADRD risk, was not associated with beta-amyloid deposition [227].
Pre-DIVA: The Pre-DIVA trial was a cluster-randomized trial that enrolled 3526 participants from 26 healthcare centers to receive either a multicomponent intervention or a usual care control group for 6-years [228]. Participants were older adults aged 70–78 and the primary exclusion was the presence of dementia. The intervention consisted of a nurse-led, cardiovascular risk management protocol with motivational interviewing techniques. In this pragmatic trial setting, incident dementia served as the primary cognitive outcome of interest, in contrast to the majority of other multicomponent RCTs that used neurocognitive performance [229]. Results demonstrated that the intervention group demonstrated improvements on multiple CVD risk markers, although notably the intervention did not reduce ‘hard’ CVD outcomes, such as myocardial infarction or stroke. Similarly, the intervention did not appear to reduce the development of dementia, with both the treatment and control participants had a comparable prevalence of incident dementia (7%). This negative finding may have been influenced by the relatively modest level of baseline cardiovascular risk and also the high standard of care within the control group [230].
The lack of a primary finding ignores several interesting nuances within the Pre-DIVA findings, however. First, although the overwhelming number of dementia cases were attributed to AD, intervention effects were much stronger among the limited subset of participants experiencing non-AD causes of dementia (HR = 0.37 [0.18, 0.76]), including VaD (HR = 0.43 [0.17, 1.12]). In addition, the authors conducted several sensitivity analyses in order to determine whether intervention may have conferred benefits among participants who 1) followed the intervention more closely and 2) had a less severe history of CVD or CVD risk at baseline. These analyses, although post hoc, seem to suggest that greater risk reductions may indeed be beneficial for participants with less advanced CVD at study entry. For example, in a per protocol analysis the treatment group showed a trend towards reduced dementia development (HR = 0.78 [0.58, 1.04]). The trend for a treatment benefit was stronger among individuals with untreated HTN (HR = 0.69 [0.43, 1.11]), particularly among those with higher levels of compliance (HR = 0.54 [0.32, 0.92]). A similar magnitude of benefit was observed when analyses were limited to participants free from a history of CVD events at baseline and who complied with the intervention (HR = 0.64 [0.44, 0.94]).
MAPT: The multidomain Alzheimer preventive trial (MAPT) was a 3-year, multicenter RCT among 1680 community-dwelling older adults aged > 70 [231]. Additional symptom-related inclusion criteria were spontaneously reporting a memory concern to their physician, limitations in one IADL, or slower gait speed. The multidomain intervention consisted of physical activity, nutrition, cognitive training, and three preventive consultations. Participants were randomly assigned to either the multidomain intervention plus omega 3 polyunsaturated fatty acids (800 mg docosahexaenoic acid and 225 mg eicosapentaenoic acid), multidomain intervention plus placebo, omega 3 polyunsaturated fatty acids alone, or placebo alone. Physical training recommendations provided to participants in MAPT were to perform at least 150 minutes of moderately intensive physical activity per week (5 sessions, 30 mins each) [232]. Recommendations were individually tailored and participants could select either aerobic exercises or strength training. The primary outcome was change from baseline to 36 months on a composite z-score combining four cognitive tests, comprised of two memory indices, ten MMSE items, the Digit Symbol Substitution Test, and Category Naming Test. There were no significant differences in 3-year cognitive decline between any of the three intervention groups and the placebo group, with the exception that the combined treatment group showed improvements on the MMSE relative to placebo. In exploratory sensitivity analyses, participants with a higher risk of dementia at baseline in the combined treatment group demonstrated modest improvements relative to other participants, with modest effect sizes on the order of treatment benefits observed in FINGER (ES = 0.131).
ADDITION: The ADDITION trial was a multinational, cluster-randomized trial in with individuals with type-2 diabetes (T2DM) were randomized to lifestyle advice for metabolic function vs. routine care [233]. Although the trial incorporated 498 participants, less than 200 had baseline neurocognitive assessment, and, within the ADDITION pragmatic design, the ‘baseline’ assessments occurred several years after initiation of the study intervention. Using a stepwise treatment process, lifestyle advice (including physical activity and diet) was implemented as a first-line treatment with integration of pharmacological treatment (e.g. statins for cholesterol or beta-blockers for hypertension) at subsequent steps if CVD risk treatment targets were not achieved. The trial utilized an extensive, 90-minute neurocognitive assessment batter with multiple tests of learning / memory, attention / processing speed, and executive function. Results of the primary trial suggested that T2DM screening was not associated with reduced CVD outcomes and both treatment and control participants exhibited weight loss, without significant between-groups differences [234, 235]. In the subgroup of participants with neurocognitive testing, all participants exhibited improved blood pressure, insulin sensitivity, and cholesterol, with only cholesterol differing between groups. Perhaps not surprisingly, neurocognitive outcomes also were not differentially improved between groups, with a slight decline in performance across domains.
ENLIGHTEN: The recently published ENLIGHTEN trial examined the impact of aerobic exercise, the DASH diet, and their combination on neurocognition among individuals with vascular cognitive impairment, no dementia (vCIND) [142, 236]. The trial used a 2X2 factorial design, with aerobic exercise and the DASH diet serving as factorial treatments, in order to examine the potentially additive benefits of both lifestyle components. Participants included sedentary, older adults (≥55), with CVD risk factors and some evidence of neurocognitive impairment based on screening battery. Aerobic training was supervised for the first three months of treatment, with participants exercising three times per week at a level of 70–85% of their peak heart rate reserve. The latter three months of exercise were performed by the participants individually, with weekly documentation of their adherence that was reviewed by study personnel. The DASH diet intervention was provided by a nutritionist in a series of 30-minute sessions, conducted weekly for the first three months and biweekly from 4–6 months. Following 6-months of treatment, participants in the exercise factor groups exhibited improved fitness, participants in the DASH factor groups exhibited improved dietary patterns, and all intervention groups demonstrated a general reduction in CVD risk factors. Examination of neurocognitive changes revealed that both aerobic exercise (ES = 0.32) and DASH diet (ES = 0.30) factor groups experienced improvements in executive function and a modified clinical dementia rating scale. Examination of treatment mechanisms revealed that greater increases in aerobic fitness, reductions in composite CVD risk, and reduced dietary salt intake were the strongest correlates of improved neurocognition.
ENCORE: The ENCORE trial examined the effects of a comprehensive weight loss intervention using aerobic exercise (for weight management) and the DASH diet, or the DASH diet alone, on changes in blood pressure and metabolic outcomes [237, 238]. The study enrolled 144 overweight / obese, middle-aged and older adults with hypertension (mean age 52 years [SD = 10]), who were not taking antihypertensive therapy. Participants exercise training consisted of supervised weekly sessions, three times per week for 30 minutes, at an intensity of 70–85% of their initial heart rate reserve. The DASH diet condition participated in an initial two-week controlled feeding period and then transitioned to weekly, supervised sessions in which a nutritionist implemented the dietary intervention using both education and cognitive-behavioral strategies for weight loss. Because the trial was designed to test the effects of the DASH diet in a free-living setting, participants were not required to meet any cognitive inclusion criteria. Although the trial was designed to assess the effects of lifestyle modification on metabolic parameters, the majority of participants also underwent neurocognitive assessments both pre and post-treatment as a secondary outcome [145]. Results demonstrated that combining aerobic exercise with the DASH diet improved neurocognition on measures of executive functioning / memory / learning (ES = 0.56) and similar improvements on measures of psychomotor speed (ES = 0.48). In addition, participants in the DASH diet alone group demonstrated similar, albeit more modest improvements on psychomotor speed tests (ES = 0.44). In addition, improvements in neurocognition were associated with both increased aerobic fitness and weight loss.
PREDIMED: One of the most widely publicized and early dietary interventions to demonstrate improvements in neurocognitive outcomes is the PREDIMED trial [239]. Participants in PREDIMED included thousands of community-dwelling, older adults (men: 55–80 years; women: 60–80 years), initially free of CVD but at high vascular risk because of the presence of either type-2 diabetes or at least three major risk factors (smoker, HTN, HLD, overweight, or family history of premature CVD). The primary PREDIMED trial examined the effects of a MeDi diet intervention on CVD outcomes with three groups: MedDiet with extra virgin olive oil; MedDiet with mixed nuts, or a control group who received nutritional advice. As previously reported, the primary trial found that participants in the intervention groups exhibited improvements on screening measures of neurocognition relative to controls, including the MMSE and clock drawing test. Secondary subgroup analyses revealed even stronger benefits among participants with selected for CVD risk [113, 240]. In addition, a follow-up analysis suggested that participants in the MeDi+olive oil group had a lower risk of developing MCI relative to controls (OR = 0.34 [0.12, 0.97]), even after accounting for additional lifestyle factors, including CVD risk factors and physical activity [241].
LOOK-AHEAD: One of the largest extant weight loss trials was the Action for Health in Diabetes study (Look AHEAD), which enrolled T2DM adults aged 45–76 years of age. Additional inclusion criteria included a BMI more than or equal to 25 kg/m2 (≥27 kg/m2 if taking insulin), HbA1c less than 11% (97 mmol/mol), systolic blood pressure less than 160 mmHg, diastolic blood pressure less than 100 mmHg, and triglycerides less than 600 mg/dL [242]. participants were randomly assigned to an intensive lifestyle intervention (ILI) or a Diabetes Support and Education (DSE) control condition. The ILI included diet modification and physical activity and was designed to induce at least a 10% weight loss following one year of treatment and then to maintain this over follow-up. Intervention participants were assigned a calorie goal (1,200–1,800 based on initial weight), with less than 30% of total calories from fat (<10% from saturated fat) and a minimum of 15% of total calories from protein. The physical activity goal was more than or equal to 175 minutes per week through activities similar in intensity to brisk walking. Participants were instructed to accumulate at least 30 mins of moderate-intensity physical activity, most days of the week [243]. Participants in the control condition were invited to three group educational sessions each year. Similar to some of the other intensive lifestyle trials in which neurocognitive outcomes were collected as a secondary outcome, the first assessment of neurocognition took place 8 years after randomization on a large subset of participants.
The treatment group demonstrated substantial weight loss, much of which was maintained at the time of first neurocognitive assessment [242]. As reported in detail elsewhere, [242] the ILI group experienced an approximate 11% weight loss at year one and maintained approximately 7% weight loss through the 8-year follow-up. In contrast, the DSE group experienced a mean weight loss of around 1% at one year and 3% at the 8-year follow-up. Neurocognition was assessed using a composite score across multiple domains, including global function, verbal fluency, verbal memory, attention, executive function, and processing speed. Examination of treatment group differences, however, revealed that performance was not consistently improved between conditions, without any significant treatment group impact. There was some evidence that the intervention differentially affected overweight compared with obese participants. Among those who were overweight but not obese at enrollment (ie, 25 kg/m2≥BMI < 30 kg/m2), ILI was associated with a mean benefit for composite cognitive function of 0.276 (0.033, 0.520), compared with a deficit of 0.086 (–0.021, 0.194) among those who were obese (nominal p value for interaction: p = 0.008). In a subsequent follow-up examination, a parallel pattern of results was observed for the development of cognitive impairment, with a baseline weight by intervention effect, such that ILI was beneficial among overweight participants and harmful among those with the highest baseline weight [217].
Results of the ILI on neuroimaging markers revealed a different pattern of findings. Among a subset of participants who underwent structural brain imaging or cerebral perfusion scans, ILI generally demonstrated improvements compared to DSE [221, 244]. For example, ILI participants demonstrated a 28% lower white matter hyperintensity volume compared to DSE participants, as well as a 9% lower ventricle volume [221]. Similarly, cerebral blood flow increased among the ILI participants relative to DSE [244]. Most interestingly, these improvements did not associate with changes in neurocognition, and changes in CBF actually demonstrated a divergence in their association with neurocognition between treatment groups, with improved CBF associating positively with neurocognition among ILI participants, but not in DSE participants.
Other Multicomponent Trials: In a smaller, single-site trial, Napoli and colleagues [245] examined the impact of a weight management using diet, exercise, or both on neurocognition among 107 obese adults. Following one-year of treatment, the authors found most improvements in several neurocognitive subtests in a battery that included the modified MMSE (3MS) and several tests of executive function. Improvements in neurocognition were largest in the combined group and exercise alone group, both of which appeared to outperform the diet and control group. Interestingly, examination of the associations between improved 3MS scores and treatment mechanisms varied somewhat across treatment groups, with a tendency to suggest that fitness and metabolic parameters were most closely associated with treatment improvements. For example, in the exercise groups the strongest associations with neurocognition were observed between improved fitness, strength, insulin sensitivity, whereas for the dietary groups insulin sensitivity appeared to be most strongly associated with neurocognitive changes. Taken together, results from available multicomponent trials demonstrate an encouraging but inconsistent pattern of findings. Although these inconsistencies make a synthesis of available evidence more difficult, they also provide insights into potential mechanistic associations that can be leveraged to inform future intervention studies.
Putative Mechanisms: Opportunities for Optimization
Mechanistic pathways linking physical activity, dietary consumption, and neurocognitive outcomes have been postulated across multiple levels within the peripheral and central nervous system. Despite their heterogeneity, increasing evidence suggests that disparate risk factors may have overlapping effects through ‘convergent pathways’ of risk, including cardiovascular, metabolic, and inflammatory systems. Conceptually, risk factors for neurocognitive impairment can be thought of as exerting their influences on the brain by affecting structural neuropathological features within the brain (e.g. cortical atrophy), compensatory factors buffering the CNS from age-associated injury (e.g. cerebrovascular reserve), and more direct effects on neurochemical messenger systems within the CNS (e.g. monoamines) (Fig. 2). Individual mechanisms are reviewed briefly below, with a focus on identifying convergent pathways of risk.
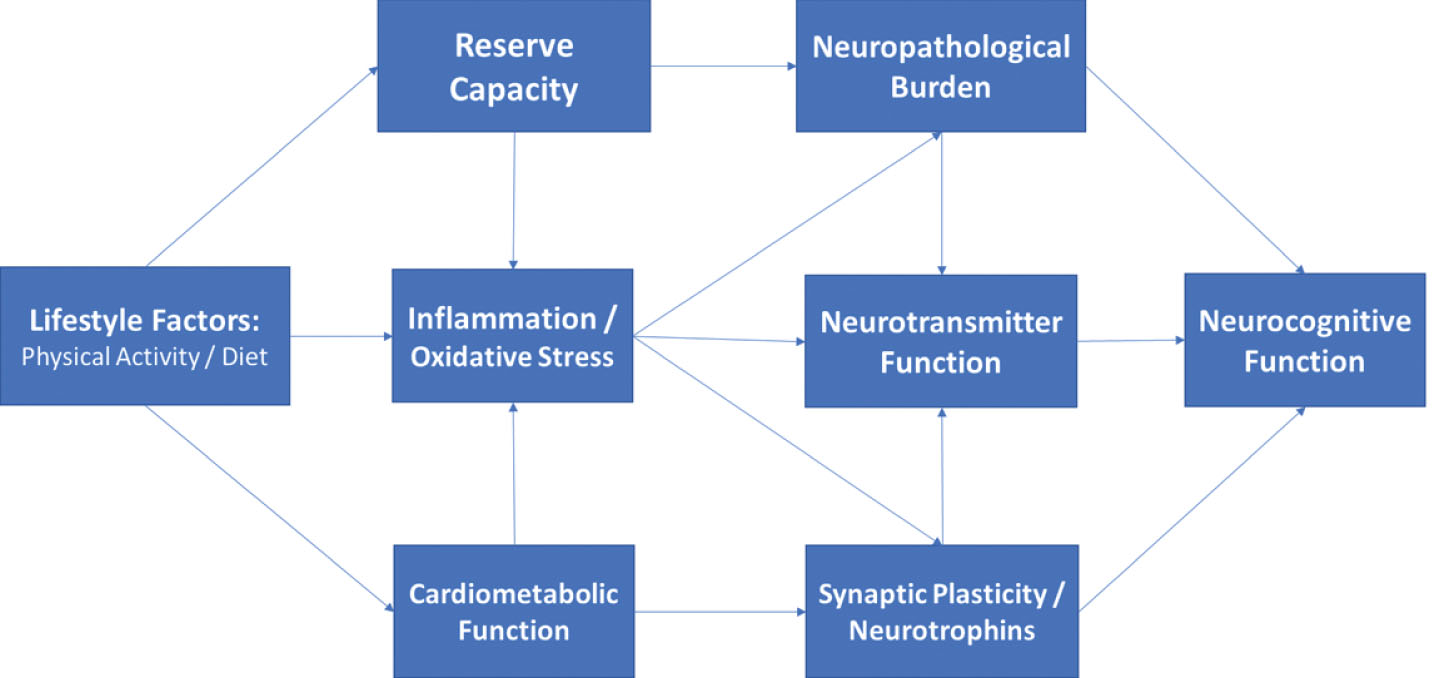
Fig.2
Directed acyclic graph linking lifestyle to neurocognition among middle-aged adults through indirect, downstream pathways including metabolic function, inflammation, and neurotrophic factors. As shown, these factors likely impact neurocognitive function through their downstream impact on brain structure and reserve capacity, with additional, indirect influences through modulation of neurotransmitter systems (e.g. monoamines).
Neurotrophins / Growth Factors
Growth factors are groups of proteins or neurohormones with effects that serve to facilitate neuronal and cerebrovascular growth, including nerve fibers, dendritic density, and arterioles [246, 247]. The primary growth factors most consistently linked to lifestyle changes are BDNF, [248–250] VEGF, [251, 252] and IGF-1, [251, 253–255] all of which are enhanced by lifestyle modification but act through distinct mechanistic channels to improve neurocognition. BDNF has been shown in numerous animal and human samples to be increased following aerobic exercise training, [256–258] for increased BDNF to associate with neurogenesis in the dentate gyrus and subventricular zone, [259–261] and for higher BDNF levels to associate with small but consistent structural increases in hippocampal volume [260]. In multiple animal studies and an increasing number of human trials, these increases in BDNF associate with improved neurocognition, particularly on tests of learning and memory [260]. In addition to these intermediate training effects, occurring over weeks and months, acute exercise has also been shown to confer transient increases in BDNF measured from peripheral sources for several hours [43, 262]. This may explain the dual effects of BDNF as having both an acute impact, such as observed among children with ADHD, and a chronic training impact in adults, both of which appear strongest on frontal-subcortical tasks.
VEGF has also been associated with lifestyle modification and is most closely tied to vascular and inflammatory functioning [263]. Endothelial cell functioning is one of the earliest, subclinical markers of vascular dysfunction, observable even in samples of obese children [263–265]. VEGF has been shown to play a critical role in angiogenesis within the brain, potentially offsetting normative damage to the small arteries with little redundancy in dorsal aspects of the prefrontal cortex. Moreover, as a marker of endothelial cell function, VEGF holds importance as a systemic, preclinical markers of vascular integrity [266]. In our own data and others, peripheral markers of endothelial function appear to mediate the associations between cerebrovascular risk factors and neurocognitive deficits in middle-aged, preclinical samples [267, 268]. In addition, endothelial cell functioning serves an important role in maintaining blood brain barrier permeability, [265, 269] which is critical for protecting the CNS from infection.
Insulin like growth factor (IGF-1) is a neurohormone with both direct and indirect influences on neurocognitive functions, primarily induced through exercise training [270, 271]. IGF-1 appears to have important regulatory effects on both BDNF and VEGF, and may therefore have a critical moderating influence on other growth factor influences within the CNS [251]. IGF-1 also appears to have independent effects on neuronal signaling, neuroprotection, and modulation of neuroinflammation [272]. More generally, IGF-1 has been hypothesized to play a mechanistic role linking peripheral impairments in metabolic function to CNS metabolic perturbations [273, 274]. IGF-1 has also been associated with neurocognition, [275] is responsive to exercise training, [276] and has been suggested to mediate the beneficial effects of exercise on brain outcomes [251].
Neuroinflammation / Oxidative Stress
Neuroinflammation is increasingly recognized as having adverse effects on brain [277, 278]. Greater levels of circulating neuroinflammatory markers are frequently shown among individuals with ADRD compared to controls, [272, 279, 280] associate with worsening neurocognition in prospective studies, [281, 282] and are upregulated among middle-aged adults with CVD risk factors and obesity [283, 284]. Neuroinflammation likely acts through several indirect pathways to worsen brain outcomes. First, the presence of elevated inflammation appears to exacerbate transient brain insults due to ischemia or hypoxia, [285] causing a prolonged recovery with potentially lingering adverse effects. Second, several lines of evidence suggest that neuropathological changes in the brain, such as beta-amyloid deposition, may independently increase inflammation levels, suggesting that they may reflect a proximal marker of neuropathological burden [279, 286]. And third, higher levels of inflammation appear to impair neurotransmitter functions, particularly in reward system brain regions critical for executive function. Taken together, elevated neuroinflammation appears to have an amplifying effect, interacting with other mechanistic channels to set of a feedback loop.
A closely related mechanism by which dietary modification may improve brain function is oxidative stress [45, 287]. Free radicals and other markers of oxidation increase with aging and are widely associated with brain health and neurocognition, with some investigators postulating that oxidative damage precedes broader neuroinflammatory changes [288]. Multiple nutritional components have been hypothesized to have beneficial effects on the brain by counteracting oxidative damage, particularly antioxidants such as vitamins A, C, and E [144, 289].
Metabolic dysfunction
A large and extensive literature base demonstrates that greater metabolic dysfunction is associated with increased ADRD risk [290, 291]. Recent consensus statements by the NIA, NINDS, and others have all advocated an increased focus on ADRD prevention, primarily focusing on three overarching risk factors: physical inactivity, ‘Western’ dietary patterns (e.g. high intake of saturated fat and complex carbohydrates, and low intake of fruits and vegetables), and poorly controlled CVD, [109, 292, 293] all of which strongly influence metabolic function. The presence of common CVD risk factors, particularly HTN and obesity, have a robust and adverse impact on neurocognitive outcomes. Obesity is one of the most common risk factors for ADRD, affecting nearly half of adults in the United States, [294, 295] and is associated with neurocognitive impairments, cortical atrophy, [76, 296, 297] and a 2-3 fold increase in long-term ADRD risk [76, 294, 298]. Midlife obesity, for example, has been associated with substantially elevated risk of impaired neurocognitive function and ADRD in later life [76, 299–302] and appears to accelerate the aging process, independent of other CVD risk factors [76]. Similar associations have been observed for HTN, with prospective studies demonstrating that the presence of HTN in midlife is associated with more than double the risk of neurocognitive decline [303–305] and dementia, [305–309] and may have particularly deleterious effects in the presence of obesity [310, 311]. As reviewed in detail elsewhere, impaired peripheral metabolic function likely impairs central metabolic function over time through insulin and leptin resistance,[50, 312] resulting in blunted CNS metabolic responses, energy exchange, and lipid metabolism.
Improving metabolic function has been hypothesized to mediate neurocognitive improvements following lifestyle modification [34, 88]. Although exercise and dietary modification both improve metabolic function, improvements in metabolic function are most closely associated with intentional weight loss, which may explain why extant studies have demonstrated improved neurocognition following variable intervention modalities, including caloric restriction [178], resistance training, [131] and aerobic paradigms [313]. This could also potentially explain why there appear to be additive effects of both aerobic training and dietary modification on neurocognitive outcomes [145, 236, 314]. Intentional weight loss among obese, middle-aged adults is increasingly recognized as a potential method to reduce ADRD risk, regardless of whether it is achieved through routine physical activity, healthier dietary patterns, or caloric restriction. For example, meta-analytic studies of middle-aged adults have demonstrated a dose-response relationship between greater weight loss and improved neurocognition, with the largest improvements observed in trials using exercise and diet to reduce weight [315]. It has been hypothesized that midlife weight loss may improve neurocognition by augmenting ‘cross talk’ between peripheral and central metabolic function, in which improved peripheral metabolism increases the efficiency and recruitment of central glucose resources and transport across the blood brain barrier [316]. Obesity is known to impair both glucose and lipid homeostasis, causing central glucotoxicity and impaired CNS insulin signaling, and weight loss may ‘recalibrate’ central homeostatic function by altering CNS metabolic mechanisms [47, 317]. Available evidence therefore indicates that pluripotent interventions “with combinatorial neuroprotective and ‘eumetabolic’ activities” [318] confer the largest neurocognitive benefits [318].
Neurogenesis / Synaptogenesis
Neurogenesis, the growth of new neurons and other neural tissue, represents one potential central pathway through which lifestyle modification improves brain function and potentially offsets normative, age-dependent brain volume loss. Synaptogenesis, the growth of new synaptic connections between neurons, is closely related to the creation of neuronal tissue [319], as immature granule cells are particularly plastic due to their sensitivity to long-term potentiation [67]. Synaptic plasticity is also a plausible downstream mechanism linking neurotrophic and metabolic factors to neurocognition, as the creation of new synaptic connections appears to be dependent both on growth factor expression and adequacy of central metabolic resources [320].
Although neurogenesis and synaptic plasticity are widely acknowledged to be improved through aerobic exercise, [32, 321] aerobic modification in and of itself appears to be a necessary but not sufficient component of neuronal growth. Although new stem cells are created, they take several weeks to integrate into the surrounding neural system and many new stem cells die off before becoming mature. The successful growth of new neurons, much like the growth of plant life in a garden, is ultimately dependent on several critical elements necessary for growth within the brain: energy, nutrients, and a conducive environment. In a simplistic model within the human brain, similar elements appear to be necessary to support neurogenesis: metabolic function, [322–324] neurotrophins, [325] and low levels of neuroinflammation [326, 327].
Several other neuropathological features of adult neurogenesis may explain their importance to the lifestyle and neurocognitive decline association. It is notable that the two primary areas of adult neurogenesis in mammals are in close proximity and have interconnections to neurocircuitry critical for learning/memory and executive functions: the dentate gyrus (proximal to the hippocampal gyrus) [259, 328] and the subventricular zone (proximal to the caudate nucleus) [329–333]. As noted earlier, these are also the two primary brain circuits implicated in AD and vascular cognitive impairment, respectively. The earliest neuropathological changes associated with AD occur in peri-hippocampal gyrus, whereas the earliest manifestations of subclinical microvascular disease are most commonly observed in periventricular brain regions because of their vulnerability to ischemia. This data could be interpreted as suggesting that the areas most vulnerable to common neuropathological changes are also most amendable to buffering and repair through behavioral modification. In addition to their impact on neurogenesis, impaired metabolic function and elevated inflammation / oxidative stress have been shown to have direct, adverse effects on neurotransmitter systems, which are worsened in the presence of both factors [334]. Higher levels of inflammation appear to disrupt connectivity between the ventral and dorsal striatum and the ventromedial prefrontal cortex [335, 336]. Both the ventral striatum and vmPFC have significant mesocorticolimbic dopamine innervations [335, 336].
Taken together, there remains a lack of consensus as to whether the effects of physical activity and dietary modification on ADRD risk overlap, are additive, or synergistic [142, 143, 214, 337–340]. For example, treatment implications would differ if the effects of physical activity and diet were found to act through aerobic fitness and inflammation, respectively, instead of both contributing to improved metabolic function. In contrast, finding synergistic effects might suggest that multicomponent interventions with both exercise and dietary components may be necessary to achieve ADRD risk reduction [129, 341–344]. Nevertheless, the present literature base suggests several important themes that may help to further optimize lifestyle approaches to ADRD prevention. First, lifestyle appears to impact the brain through pluripotent mechanisms, which vary in their importance across chronic and acute intervention settings. Second, many peripheral and indirect pathways connecting lifestyle to the brain have convergent, downstream mechanistic pathways, including central metabolic function, growth factor modulation, neuroinflammation, and neurotransmitter functions. And finally, the effects of lifestyle seem strongest when implemented early, with the greatest potential to prevent ADRD seeming to be in middle-age. Evidence increasingly may therefore suggest a ‘critical window’ for intervention among middle-aged individuals with ADRD risk factors, identified before compensatory cerebrovascular remodeling, systemic metabolic dysfunction, [345, 346] or elevated neuroinflammation set in motion a cascade of changes that may require more than lifestyle change to treat [347–350].
Mechanistic assumptions and potential solutions
“Explanations exist; they have existed for all time; there is always a well-known solution to every human problem — neat, plausible, and wrong.” [351]
“[Many] arguments are kept alive by a failure to acknowledge nuance... to argue one point using two entirely different sets of assumptions, like two tennis players . . . hitting beautifully executed shots from either end of separate tennis courts.” [352]
Despite the overwhelming amount of evidence linking variations in lifestyle to differential brain outcomes from both animal and human literatures, there remains a lack of consensus as to whether lifestyle modification represents a viable treatment approach to mitigate ADRD risk among middle-aged and older adults. One could argue from the present review that exercising regularly and eating a healthier diet can clearly improve brain and neurocognitive function among some individuals. The most pressing questions are therefore less concerned with whether lifestyle modification can improve brain function but how and for whom. These latter questions can be informed by contemporary analytical and methodological advancements designed to address these specific issues: causal inference analytical paradigms [353, 354] and optimization trial designs [355, 356]. These techniques can readily be applied to several, potentially implicit assumptions, regarding the causal nature and underlying mechanisms linking exercise, diet, and the brain. Testing these assumptions may provide critical insights to advance ADRD preventive efforts.
Assumption 1: Homogeneous Mechanisms of Cause and Cure
Put simply, the causes and cures of neurocognitive impairment may not be the same, suggesting that different treatment approaches may be needed beyond those specifically targeting the causes of initial neurocognitive change. This may be one reason why simply reducing CVD risk factors does not consistently improve neurocognition: the presence of CVD risk causes a cascade of CNS damage that is only partially reversible once the risk factors are mitigated. The distinction in mechanisms can also be seen by a brief examination of their time-courses, with both acute and chronic effects observed [357]. For example, while the adverse effects of hypertension (HTN) on brain function are widely accepted, [358] the downstream mechanisms by which HTN impairs brain function exert their influence over the course of decades, [53, 359] whereas lifestyle HTN trials demonstrate benefits over the course of several months, [145, 236]. Moreover, reductions in white matter hyperintensities may take years to accrue [221] and do not necessarily associate with neurocognitive improvements from available trial data [242].
Assumption 2: Mechanisms are Monolithic
As easily seen in the available animal literature, many of the proposed mechanisms linking lifestyle modification to neurocognition interact with other downstream mechanistic pathways. For example, metabolic function may interaction with inflammation to increase ADRD risk [360]. While multiple mediators are widely acknowledged in animal literature, available lifestyle interventions are strikingly simplistic in their analysis of treatment mechanisms from available trials. Contemporary analytical approaches now incorporate approaches for examining multiple mechanisms and even interactions between mechanisms, which could plausibly be hypothesized to impact brain function [361, 362]. For example, the association between increased BDNF and improvements in learning might be moderated by inflammation levels, increasing learning for those with low inflammation but failing to do so among individuals with high levels. Similarly, it is possible that neurocognitive improvements may be mediated by parallel mechanistic processes, by interacting mechanistic processes, or by different mediators for different individuals. Because lifestyle modification is pluripotent, all of these scenarios are plausible and testable within contemporary analytical paradigms [363, 364]. Moreover, examination of multiple mechanisms could potentially explain the lack of a main effect in some cases, if one mechanistic process has opposing effects through a separate mechanistic pathway, [363] or if the association between the putative mediator and the outcome is confounded by an unmeasured factor. In the HTN example above, for example, the effects of BP reduction on neurocognition could plausibly confounded by impaired cerebral autoregulation or cerebrovascular reserve capacity, which are blunted by chronically elevated HTN and are not conventionally accounted for or even assessed.
Although the notion that mechanisms may interact may seem unnecessarily complex, it is consistent with several aspects of the available literature on exercise and neurocognition, which has reproducibly demonstrated that aerobic exercise training improves neurocognition, [117, 118, 365] but failed to find any association between improved fitness and neurocognitive improvements [133]. Although the magnitude of treatment effect has varied, most meta-analyses of exercise interventions and cognitive function have reported improvements across multiple domains of function [129, 366, 367]. Surprisingly, the associations between putative mechanistic features within these trials and cognitive improvements has been far less consistent, with the majority of evidence from systematic reviews of both older [136] and more contemporary trial data failing [133] to find a dose-response association between improved aerobic fitness and improved cognitive outcomes. Observational data can be interpreted as showing a similar, non-linear effect, such that modest to moderate levels of activity are most critical for improving cognitive outcomes, with diminishing returns beyond this threshold [112, 135]. This could suggest that improvements in neurocognition may parallel improvements in metabolic function, which have been shown to have a threshold effect in response to various training paradigms and may not mirror aerobic improvements [368, 369].
Assumption 3: Uniformity of the Mediator-Outcome Association
Another critically important and often overlooked assumption is that putative mediators of treatment act uniformly across participants. In other words, if one hypothesizes that improved fitness will improve neurocognition, they may also implicitly assume that this association is similar enough across participants to use linear regression-based approaches. Unfortunately, contemporary causal inference approaches have demonstrated this is often not the case for several reasons: 1) the mediating variable is not normally distributed, 2) the mediator interacts with the treatment variable to differentially modify outcomes (treatment-exposure interaction), or 3) the association underlying changes in the mediator and changes in outcome is confounded by a related but unmeasured confounder [354, 370]. One has to look no further than the LOOK-AHEAD trial to find evidence for a treatment-exposure interaction, with the effects of weight loss on neurocognition varying across participants [242], as well as the association between improved CBF and neurocognition showing differential associations across participants [244]. These findings suggest that unmeasured factors likely impacted the mechanistic function of weight loss, perhaps with more metabolically compromised participants experiencing destabilized central metabolism in the presence of weight reduction.
It is likely that treatment-exposure interactions explain some of the disparate findings across trials, as well as the potential presence of a ‘tipping point’ beyond which lifestyle modification does not appear to improve neurocognitive outcomes. Although the precise causes of these large individual differences are unclear, they appear to vary based on two premorbid characteristics that differ widely across patient populations: degree of metabolic impairment and neuropathological burden. Meta-analyses of lifestyle intervention trials have reported age-dependent effects on neurocognition, with larger benefits among middle-aged and older adults free from cognitive impairment, [36, 117, 128, 129, 371] suggesting a metabolic ‘tipping point’ for intervention efficacy [365, 366, 372]. In addition, trials enriched for metabolic disturbances appear to have a greater degree of individual treatment variation, with more metabolically compromised individuals exhibiting lesser treatment improvements, or in some cases showing an adverse impact. Paradoxically, later behavioral intervention among individuals with systemic vascular dysfunction may actually precipitate cognitive decline, [215, 220, 306, 373, 374] despite the beneficial effects of CMRF management at early disease stages [10, 375–377]. This could explain why recent multicomponent trials among participants with impaired metabolic function, including the Diabetes Prevention Program (DPP) [378] and LOOK-AHEAD trials, found that behavioral weight loss does not necessarily improve neurocognition [215–219] and may even have deleterious effects in more metabolically compromised individuals, [220] despite improvements in some neuroimaging biomarkers [221, 222].
Similar individual differences have been noted for CVD risk reduction, particularly for hypertension. The presence of CVD risk factors in midlife is one of the most widely documented and robust risk factors for future development of ADRD [379, 380]. Interestingly, the association between CVRFs and ADRD appears age-dependent, with midlife CVRFs drastically increasing risk, whereas the presence of CVD risk factors in older adults having equivocal or even protective associations with ADRD development [85, 381, 382]. It is possible that these disparate associations result from unmeasured individual differences in metabolic or brain reserve capacities, with chronic exposure to CVD risk compromising the ability of the scaffolding / reserve system to withstand additional age-related insults. Because chronic engagement in healthy lifestyle habits may reduce ADRD through its parallel effects on cognitive and metabolic reserve capacity, [89, 96, 383, 384] collection of more detailed historical information on midlife lifestyle habits may be informative, even among trials in older adults, because of its potential function as an unmeasured moderator of the treatment-exposure association.
Individual differences in reserve capacities may also explain large variations in treatment-related cognitive outcomes from randomized trials of exercise [19, 36, 116, 128, 365, 366], similar to those observed following intentional weight loss [186, 215, 217, 220, 315, 385].
Assumption 4: All Neuropathological Changes are Created Equally
Although a comprehensive discussion of functional neuroanatomy is beyond the scope of the present review, it is worth noting that the maintenance of some higher order neurocognitive functions is preferentially dependent on neurocircuitry in the frontal lobe and FSCs [386, 387]. While these areas generally undergo uniform, age-associated degradation through middle-age, occult microvascular damage and cortical atrophy are endemic among older adults, with substantial variation [388–394]. It is widely documented that strategic infarcts to the caudate nucleus, basal ganglia, and other critical brain circuits can cause precipitous cognitive impairment leading to dementia in a normally functioning adult under some circumstances [376]. Although rare in their pure form, [395] the conceptual relevance of disconnectivity between functionally critical brain circuits and larger regions is well-known and increasingly implicated as important for ADRD risk [396, 397]. In contrast to the known functional neuroanatomical correlates of intact neurocognition, most lifestyle RCTs examining mechanistic data have relied on global markers of brain health (e.g. cortical atrophy, microvascular burden), which may obscure wide variation among older participants. This is particularly important for examinations of lifestyle and neurocognitive dysfunction, which may only become manifest after substantial neuropathological burden has accrued.
The potential importance of a circuit-based appreciation for neurocognitive change is particularly relevant within the aerobic exercise literature, for which treatment improvements are preferentially observed on tests of frontal-subcortical function and more broadly in the ‘executive control’ (ECN) and ‘salience’ networks (SN), as well as their reciprocal associations with the ‘default mode’ network (DMN), all of which work in concert to facilitate complex neurocognitive functions. Within these networks, fronto-subcortical brain regions (e.g. anterior cingulate cortex, ventromedial prefrontal cortex, insular cortex) [398] interact reciprocally with critical ‘hub’ regions in the posterior cingulate cortex involved in the DMN [399]. DMN activity is traditionally anti-correlated with task-based networks, such as the SN [400, 401]. Thus, coordination of the DMN and SN are critical to attentional / executive control and reduction of internal distractors [402, 403]. A circuit- or network-based interpretation of aerobic exercise effects would synthesize mechanistic studies demonstrating that physical activity improves functioning within FSCs across disparate patient populations, including older adults, [117, 404] vCIND, [73, 236] Parkinson’s Disease, [405, 406], bipolar, [407] and ADHD [75].
Areas within the SN and ECN are also critical for executive functions because of they have a greater density of ‘rewards system’ neurotransmitter receptor sites, particularly for monoamines such as noradrenaline and dopamine. Noradrenergic function, in particular, is increasingly recognized as having an important role in the development of cognitive impairment among older adults and is critical for the maintenance of frontal-lobe functions [408, 409]. Moreover, these findings have recently been extended to healthy aging samples, with multiple investigators proposing that NE activation facilitating by the locus coeruleus may represent the best central nervous system biomarker of cognitive reserve (i.e. LC-reserve hypothesis) [410, 411]. Although no adult trials, to our knowledge, have examined the potentially moderating role of catecholamines, this association has been demonstrated in adolescent samples [412].
Assumption 5: Large, Randomized Trials are Necessary to Delineate Specific Treatment Mechanisms
Examination of the available literature demonstrates that most of the available trial data on neurocognitive outcomes comes from large, exceedingly costly trials in which multicomponent treatment packages were typically ‘bundled’ into one group. For example, the FINGER trial utilized four different intervention components: diet, exercise, CVD risk management, and cognitive training [214]. Although RCTs will ultimately carry the greatest scientific significance to change clinical practice, and emerging wave of multiphase optimization strategy (MOST) trials have provided critical insight into some recalcitrant public health problems [355]. Using an elaborated factorial design, particularly sequential multiple assignment randomized trials (SMART), these approaches allow investigators to systematically test treatment sequencing, optimize intervention components, and disentangle treatment-exposure interactions systematically using an efficient design with small sample size requirements. SMART designs in particular are ideal for patient populations with large individual differences in treatment response, with a secondary randomization occurring based on initial treatment response. For example, a MOST-based design in which exercise, diet, and cognitive training was examined would be helpful in determining their independent and synergistic effects (Table 1).
Table 1
Example of a factorial design to determine optimal intervention components among exercise, diet, and cognitive training. Within the example below, main effect comparisons for factors would consist of 1) Exercise vs. No-Exercise (conditions 1, 2, 3, 4 vs. 5, 6, 7, 8); 2) Diet vs. No-Diet (conditions 1, 2, 5, 6 vs. 3, 4, 7, 8); and Cognitive training vs. No-cog training (conditions 1, 3, 5, 7 vs. 2, 4, 6, 8). Interactions between intervention factors can also be examined
| Experimental Condition | Exercise Factor | Diet Factor | Cognitive Training Factor |
| 1 | Exercise | Diet | Cog Training |
| 2 | Exercise | Diet | No-Cog Training |
| 3 | Exercise | No-Diet | Cog Training |
| 4 | Exercise | No-Diet | No-Cog Training |
| 5 | No-Exercise | Diet | Cog Training |
| 6 | No-Exercise | Diet | No-Cog Training |
| 7 | No-Exercise | No-Diet | Cog Training |
| 8 | No-Exercise | No-Diet | No-Cog Training |
MOST and SMART-based approaches could be adopted for brain-based mechanisms for which multiple treatment elements must be integrated in order to improve outcomes. SMART design approaches could be used to test, for example, whether training modalities that first target inflammation or metabolism before introducing other treatment elements may have the greatest benefits, if these treatment mechanisms truly do block other downstream brain changes. These kinds of approaches appear particularly relevant to test the effects of cognitive training in conjunction with lifestyle modification, [413] as cognitive engagement is increasingly recognized as a pillar of prevention [173, 414] and may have contributed to the observed success of the FINGER trial [213, 224, 415]. It is possible that discrepant findings between FINGER and other combined interventions, such as the MAX trial, [416] could have been that elements of the dietary intervention served to ‘prime’ brain areas needed for cognitive improvements for plasticity by augmenting metabolic or inflammatory pathways. Such sequential approaches have been hypothesized to confer greater cognitive benefits among older adults in cognitive rehabilitation [413].
Future directions
As shown by the present review, the impact of lifestyle on the brain, as well as the field of ‘health neuroscience’ more broadly, [417] is a burgeoning area with a great diversity of avenues for future inquiry. This is particularly relevant as intervention strategies are increasingly personalized for improved treatment effect [418]. Several specific hypotheses could be tested as an extension of the associations reviewed above. First, because neuropathological changes in the brain occur over the course of decades and may vary widely between individuals, [55] efforts to assess and better understand individual differences in chronicity of disease burden and structural brain imaging markers on neurocognitive changes following lifestyle change appear indicated. Future interventions would also be informed from inferences brought about by the NIA’s task force on ‘cognitive reserve’, [86, 94, 96] so that randomized trials can incorporate codified markers of reserve capacity to either examine as modifiers of treatment response or at least as a individual difference variable to control for in their examination of treatment changes [419]. Moreover, future interventions should consider inclusion criteria that partially account for reserve factors, such as the use of demographically-corrected normative data, which provide deviations in neurocognitive performance based both age and premorbid education level (a conventional marker of cognitive reseve). Indeed, it is notable that trials accounting for individual differences in baseline neurocognitive function, disease burden (e.g. vascular risk), and cognitive reserve capacity (e.g. education level [145, 236] or indirectly through CAIDE [214]) have demonstrated the largest treatment-related improvement in neurocognition.
Second, routine collection where possible of ‘downstream mechanistic pathways’ (e.g. metabolic function, inflammation, and brain structure) could help inform mechanistic pathways in future studies, even if other pathways are unmeasured (neurotrophins and neurotransmitter function). These would allow emerging trials to align with contemporary causal inference analytic paradigms in which ‘blocking’ of various causal pathways could prove highly informative [353, 420]. Moreover, collection of detailed historical data on cumulative exposure to CVD risk factors could provide important information on individual differences in reserve capacity, in order to examine whether this functions as an unmeasured individual difference influencing the observed treatment-exposure interactions.
A third potential extension from the present review is the need to carefully account for basal levels of inflammation, which may function as a conceptually central mechanism blocking the effects of indirect mechanistic pathways. Elevated inflammation has been shown to mitigate neurotrophic effects on neurocognition, [421] is common among individuals with CVD risk factors and obesity in particular, [278] has been associated with biomarkers of preclinical ADRD progression, [422] and has been robustly associated with neurocognitive decline [277]. This could partially explain the lack of neurocognitive improvements in some comprehensive lifestyle trials, such as LOOK-AHEAD [242]. This may also explain why individuals with more advanced neuropathological burden, such MCI or AD, may not experience the same cognitive improvements as cognitively normal older adults [372] as the presence of higher beta amyloid load has been suggested to promulgate elevated neuroinflammatory levels in a feedback loop [279, 423–425]. Future trials may therefore benefit from optimizing intervention components that are most closely associated with improved inflammation, such as weight loss and an anti-inflammatory diet, may therefore be indicated.
Fourth, the application of SMART-based designs that sequentially improve metabolic function or inflammation may provide critical insights for the sequencing of future intervention trials. For example, trials in which participants have been ‘primed’ to maximize the benefits of cognitive training could first attempt to optimize metabolic or inflammatory targets, with secondary randomization to cognitive training. Within this framework, finding that metabolic or inflammatory systems must be optimized in order to experience cognitive gains may suggest that treatments should first attempt to induce neuroplasticity as the first line of treatment. In addition to testing for sequencing effects, this type of approach would also allow for granular examination of which individuals were unable to augment metabolic or inflammatory systems before additional intervention. This would provide critically important data on which individuals may be most or least likely to respond to such an intervention approach, as well as which individuals may be able to meet intermediate treatment targets with additional intervention. Regardless of the findings, such approaches, if carefully performed, would provide clinically meaningful information to inform prevention efforts using personalized approaches.
Although SMART-based approaches have not generally been utilized in geriatric samples, likely because of the relative insensitivity of cognitive tests to detect early treatment improvements, several paradigms could be considered leveraging such approaches. For example, although early markers of treatment response have not been systematically used in geriatric samples, several conventions have been explored and could be investigated more rigorously for personalized treatment approaches. Short-interval biomarkers of improvement have been published both for neurocognitive function itself, [426] changes in IADLs based on functional assessment, [427] as well as putative mechanistic markers of brain reserve, including surface-based cortical thickness indices [428, 429] and mesial temporal lobe morphological features [430–433]. Because these morphological features experience accelerated change in some clinical samples, such as MCI, [431], and are impacted by lifestyle factors [43, 260] and vascular risk, [434] it is plausible they could be examined as intermediate markers of treatment response. Although less sensitive to treatment improvements, available markers appear to hold promise for testing intervention effects on stabilization of function in populations hypothesized to be experiencing a progressive decline.
SMART-based designs also have several advantages over conventional treatment approaches in their ability to test treatment response to unimodal vs. multimodal interventions targeting ‘downstream’ mechanistic factors (e.g. metabolic function), as well as treatment sequencing for potential ‘priming’ effects. For example, because interventions impacting metabolic function may be particularly important for neurocognitive gains, a natural extension may be investigating which individuals benefit from conventional exercise training modalities (e.g. brisk walking or jogging) vs. aerobic training with high intensity interval training, which more directly targets metabolic parameters (Fig. 3). Additional dietary modification and/or caloric restriction could also be employed for non-responders as a means of further metabolic enhancement. In addition to testing the main effects of primary intervention, this also provides rich clinical data to characterize individuals factors associated with initial treatment response, such as older age, greater metabolic dysfunction, vascular risk, and APOE genotype. Cognitive training interventions could also be employed with or without exercise training (Fig. 4). In addition to testing the additive benefits of exercise, such designs would allow for testing whether treatment improvements emerge after longer treatment duration, whether behavioral modification to induce neuroplasticity is needed to enhance cognitive training, and characterizing individual differences in initial treatment response.
Fig.3
Example of a SMART-based design among older adults with metabolic syndrome. In this example, participants are initially randomized to either walking or a more metabolically intensive intervention using high intensity interval training (HIIT). The tailoring variable used to determine secondary randomization is stabilization of cortical atrophy. Secondary randomization among non-responders introduces an additional metabolic treatment, dietary modification, in order to determine if some individuals are able to achieve improved cognitive outcomes when metabolic parameters are targeted with additional treatment components.
Fig.4
Example of a SMART-based design among older adults with mild cognitive impairment (MCI). In this example, participants are initially randomized to either cognitive training or a combined cognitive and physical activity program, that may augment neuroplasticity. The tailoring variable used to determine secondary randomization is improvements on a brief measure of processing speed, which has been associated with intervention responsivity in other trial settings. Secondary randomization among non-responders introduces an additional treatment augmenting metabolic and inflammatory function, dietary modification, in order to determine if some individuals are able to achieve improved cognitive outcomes with additional treatment.
Finally, if individual differences in background characteristics continue account for substantial variation in treatment improvements, more nuanced approaches to leverage variation from neurocognitive assessment batteries may prove beneficial. Similar applications using Bayesian modeling approaches of individual variation in neuroimaging outcomes are increasingly utilized to model the integrity of functionally integrated neurocircuitry [435–438] across structural and functional assessment modalities [439–441]. An increasingly common approach across multiple neurocognitive tests is the use of decomposition methods to quantify the level of overall function, domain-specific function, and variation across assessment measures [442–444]. Emerging evidence suggests that both the overall level of performance and variation across performance measures represent markers of cognitive reserve with unique predictive abilities [445]. Because increased inefficiencies in functional connectivity precede overall behavioral decline, [446, 447] use of decomposition methods may be particularly important among preclinical samples [448].
Conclusions
Dietary and exercise interventions hold promise for improving neurocognition, although we are just beginning to determine the optimal components, intervention modalities, and downstream mechanisms of treatment benefit. As noted in the present review, there remain large individual differences in treatment response that we are only beginning to understand. Future studies may benefit from inclusion of codified measures reflecting individual differences in chronicity or degree of disease burden and markers of cognitive reserve. Optimization strategies to determine the optimal timing and modalities of intervention hold critical importance for the development and implementation of future ADRD prevention efforts.
CONFLICT OF INTEREST
Dr. Patrick Smith has no conflicts of interest to declare. He is actively funded on an NIH trial examining the effects of dietary modification and neurocognition.
ACKNOWLEDGMENTS
Dr. Smith would like to thank Dr. Eric Daza, Ms. Stephanie Mabe, and Dr. Dan Blazer for their thoughtful input throughout the writing of this manuscript. This research was supported by funding from the National Institute of Health, NHLBI Grant # R01HL130237.
REFERENCES
[1] | Blazer DG . Cognitive Aging: What We Fear and What We Know. Perspect Biol Med. (2017) ;60: (4):569–82. |
[2] | Livingston G , Sommerlad A , Orgeta V , et al. Dementia prevention, intervention, and care. Lancet. (2017) . |
[3] | Graham JE , Rockwood K , Beattie BL , et al. Prevalence and severity of cognitive impairment with and without dementia in an elderly population. Lancet. (1997) ;349: (9068):1793–6. |
[4] | Plassman BL , Langa KM , Fisher GG , et al. Prevalence of cognitive impairment without dementia in the United States. Annals of internal medicine. (2008) ;148: (6):427–34. |
[5] | Brookmeyer R , Johnson E , Ziegler-Graham K , Arrighi HM . Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. (2007) ;3: (3):186–91. |
[6] | Hurd MD , Martorell P , Langa KM . Monetary costs of dementia in the United States. The New England journal of medicine. (2013) ;369: (5):489–90. |
[7] | Stefanacci RG . The costs of Alzheimer’s disease and the value of effective therapies. Am J Manag Care. (2011) ;17: (Suppl 13):S356–362. |
[8] | Alzheimer’s A. 2010 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia. (2010) ;6: :158–94. |
[9] | Alzheimer’s A. Alzheimer’s disease. Facts and Figures. Alzheimer’s and Dementia. (2008) ;4: (2). |
[10] | Gorelick PB , Scuteri A , Black SE , et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke; a journal of cerebral circulation. (2011) ;42: (9):2672–713. |
[11] | Barnes DE , Yaffe K . The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. (2011) ;10: (9):819–28. |
[12] | Plassman BL , Williams JW , Burke JR , Holsinger T , Benjamin S . Systematic review: Factors associated with risk for and possible prevention of cognitive decline in later life. Annals of Internal Medicine. (2010) ;153: (3):182–93. |
[13] | Tricco AC , Ashoor HM , Soobiah C , et al. Comparative Effectiveness and Safety of Cognitive Enhancers for Treating Alzheimer’s Disease: Systematic Review and Network Metaanalysis. J Am Geriatr Soc. (2018) ;66: (1):170–8. |
[14] | Cummings J , Ritter A , Zhong K . Clinical Trials for Disease-Modifying Therapies in Alzheimer’s Disease: A Primer, Lessons Learned, and a Blueprint for the Future. J Alzheimers Dis. (2018) ;64: (s1):S3–S22. |
[15] | Cummings J . Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci. (2018) ;11: (2):147–52. |
[16] | Cavedo E , Lista S , Khachaturian Z , et al. The Road Ahead to Cure Alzheimer’s Disease: Development of Biological Markers and Neuroimaging Methods for Prevention Trials Across all Stages and Target Populations. The Journal of Prevention of Alzheimer’s Disease. (2014) ;1: (3):181–202. |
[17] | Chuang YF , An Y , Bilgel M , et al. Midlife adiposity predicts earlier onset of Alzheimer’s dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol Psychiatry. (2016) ;21: (7):910–5. |
[18] | Iadecola C , Yaffe K , Biller J , et al. Impact of Hypertension on Cognitive Function: A Scientific Statement From the American Heart Association. Hypertension. (2016) ;68: (6):e67–e94. |
[19] | Lehert P , Villaseca P , Hogervorst E , Maki PM , Henderson VW . Individually modifiable risk factors to ameliorate cognitive aging: A systematic review and meta-analysis. Climacteric. (2015) ;18: (5):678–89. |
[20] | de Oliveira FF , Pivi GAK , Chen ES , Smith MC , Bertolucci PHF . Risk factors for cognitive and functional change in one year in patients with Alzheimer’s disease dementia from Sao Paulo, Brazil. J Neurol Sci. (2015) ;359: (1-2):127–32. |
[21] | Etgen T , Sander D , Bickel H , Forstl H . Mild cognitive impairment and dementia: The importance of modifiable risk factors. Dtsch Arztebl Int. (2011) ;108: (44):743–50. |
[22] | Duron E , Hanon O . Vascular risk factors, cognitive decline, and dementia. VascHealth Risk Manag. (2008) ;4: (2):363–81. |
[23] | Barnes DE , Whitmer RA , Yaffe K . Physical activity and dementia: The need for prevention trials. ExercSport SciRev. (2007) ;35: (1):24–9. |
[24] | Orgeta V , Mukadam N , Sommerlad A , Livingston G . The Lancet Commission on Dementia Prevention, Intervention, and Care: A call for action. Ir J Psychol Med. (2019) ;36: (2):85–8. |
[25] | In: Downey A , Stroud C , Landis S , Leshner AI , eds. Preventing Cognitive Decline and Dementia: A Way Forward. Washington (DC), (2017) . |
[26] | Blazer DG , Yaffe K , Karlawish J . Cognitive aging: A report from the Institute of Medicine. Jama. (2015) ;313: (21):2121–2. |
[27] | Debette S , Seshadri S , Beiser A , et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. (2011) ;77: (5):461–8. |
[28] | Alonso A , Jacobs DR Jr , Menotti A , et al. Cardiovascular risk factors and dementia mortality: 40 years of follow-up in the Seven Countries Study. J NeurolSci. (2009) ;280: (1-2):79–83. |
[29] | Davis JC , Bryan S , Marra CA , Hsiung G-YR , Liu-Ambrose T . Challenges with cost-utility analyses of behavioural interventions among older adults at risk for dementia. Br J Sports Med. (2015) ;49: (20):1343–7. |
[30] | Barnes DE , Yaffe K . The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. (2011) ;10: (9):819–28. |
[31] | Meadows DH . Thinking in Systems: A Primer. United Kingdom: Earthscan; (2008) . |
[32] | Phillips C . Lifestyle Modulators of Neuroplasticity: How Physical Activity, Mental Engagement, and Diet Promote Cognitive Health during Aging. Neural Plast. (2017) ;2017: :3589271. |
[33] | Cammisuli DM , Innocenti A , Franzoni F , Pruneti C . Aerobic exercise effects upon cognition in Mild Cognitive Impairment: A systematic review of randomized controlled trials. Arch Ital Biol. (2017) ;155: (1-2):54–62. |
[34] | Bertram S , Brixius K , Brinkmann C . Exercise for the diabetic brain: How physical training may help prevent dementia and Alzheimer’s disease in T2DM patients. Endocrine. (2016) ;53: (2):350–63. |
[35] | Barnes JN . Exercise, cognitive function, and aging. Adv Physiol Educ. (2015) ;39: (2):55–62. |
[36] | Young J , Angevaren M , Rusted J , Tabet N . Aerobic exercise to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst Rev. 2015: (4):CD005381. |
[37] | Sanders ML , Stuckenschneider T , Devenney KE , et al. Real World Recruiting of Older Subjects with Mild Cognitive Impairment for Exercise Trials: Community Readiness is Pivotal. J Alzheimers Dis. (2018) ;62: (2):579–81. |
[38] | Langoni CDS , Resende TL , Barcellos AB , et al. Effect of Exercise on Cognition, Conditioning, Muscle Endurance, and Balance in Older Adults With Mild Cognitive Impairment: A Randomized Controlled Trial. J Geriatr Phys Ther. (2018) . |
[39] | Fabbri L , Mosca IE , Gerli F , et al. The Games for Older Adults Active Life (GOAL) Project for People With Mild Cognitive Impairment and Vascular Cognitive Impairment: A Study Protocol for a Randomized Controlled Trial. Front Neurol. (2018) ;9: :1040. |
[40] | De Wit L , O’Shea D , Chandler M , et al. Physical exercise and cognitive engagement outcomes for mild neurocognitive disorder: A group-randomized pilot trial. Trials. (2018) ;19: (1):573. |
[41] | de Souto Barreto P , Demougeot L , Vellas B , Rolland Y . Exercise Training for Preventing Dementia, Mild Cognitive Impairment, and Clinically Meaningful Cognitive Decline: A Systematic Review and Meta-analysis. The journals of gerontology Series A, Biological sciences and medical sciences. (2018) ;73: (11):1504–11. |
[42] | Best JR , Chiu BK , Liang Hsu C , Nagamatsu LS , Liu-Ambrose T . Long-Term Effects of Resistance Exercise Training on Cognition and Brain Volume in Older Women: Results from a Randomized Controlled Trial. J Int Neuropsychol Soc. (2015) ;21: (10):745–56. |
[43] | Voss MW , Erickson KI , Prakash RS , et al. Neurobiological markers of exercise-related brain plasticity in older adults. Brain Behav Immun. (2013) ;28: :90–9. |
[44] | Pieramico V , Esposito R , Sensi F , et al. Combination training in aging individuals modifies functional connectivity and cognition, and is potentially affected by dopamine-related genes. PLoS One. (2012) ;7: (8):e43901. |
[45] | Sripetchwandee J , Chattipakorn N , Chattipakorn SC . Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia. Front Endocrinol (Lausanne). (2018) ;9: :496. |
[46] | Martins IV , Rivers-Auty J , Allan SM , Lawrence CB . Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer’s Disease. J Alzheimers Dis. (2017) ;55: (3):915–32. |
[47] | Pugazhenthi S . Metabolic Syndrome and the Cellular Phase of Alzheimer’s Disease. Prog Mol Biol Transl Sci. (2017) ;146: :243–58. |
[48] | Wearick-Silva LE , Orso R , Martins LA , et al. Dual influences of early life stress induced by limited bedding on walking adaptability and Bdnf/TrkB and Drd1/Drd2 gene expression in different mouse brain regions. Behav Brain Res. (2019) ;359: :66–72. |
[49] | Han TK , Leem YH , Kim HS . Treadmill exercise restores high fat diet-induced disturbance of hippocampal neurogenesis through beta2-adrenergic receptor-dependent induction of thioredoxin-1 and brain-derived neurotrophic factor. Brain Res. (1707) ;1707: :154–63. |
[50] | de la Monte SM , Tong M , Daiello LA , Ott BR . Early-Stage Alzheimer’s Disease Is Associated with Simultaneous Systemic and Central Nervous System Dysregulation of Insulin-Linked Metabolic Pathways. J Alzheimers Dis. (2019) . |
[51] | Yin H , Tian S , Huang R , et al. Low Plasma Leptin and High Soluble Leptin Receptor Levels Are Associated With Mild Cognitive Impairment in Type 2 Diabetic Patients. Front Aging Neurosci. (2018) ;10: :132. |
[52] | Pegueroles J , Jimenez A , Vilaplana E , et al. Obesity and Alzheimer’s disease, does the obesity paradox really exist? A magnetic resonance imaging study. Oncotarget. (2018) ;9: (78):34691–8. |
[53] | Bilello M , Doshi J , Nabavizadeh SA , et al. Correlating Cognitive Decline with White Matter Lesion and Brain Atrophy Magnetic Resonance Imaging Measurements in Alzheimer’s Disease. J Alzheimers Dis. (2015) ;48: (4):987–94. |
[54] | Manschot SM , Biessels GJ , de VH , et al. Metabolic and vascular determinants of impaired cognitive performance and abnormalities on brain magnetic resonance imaging in patients with type 2 diabetes. Diabetologia. (2007) ;50: (11):2388–97. |
[55] | Jack CR Jr , Knopman DS , Jagust WJ , et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. (2013) ;12: (2):207–16. |
[56] | Veitch DP , Weiner MW , Aisen PS , et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. (2019) ;15: (1):106–52. |
[57] | Salthouse TA . Trajectories of normal cognitive aging. Psychol Aging. (2019) ;34: (1):17–24. |
[58] | Tsapanou A , Habeck C , Gazes Y , et al. Brain biomarkers and cognition across adulthood. Hum Brain Mapp. (2019) . |
[59] | Salthouse TA , Atkinson TM , Berish DE . Executive functioning as a potential mediator of age-related cognitive decline in normal adults. J Exp Psychol Gen. (2003) ;132: (4):566–94. |
[60] | Du J , Xu Q . Neuroimaging studies on cognitive impairment due to cerebral small vessel disease. Stroke and Vascular Neurology. (2019) :svn-2018-000209. |
[61] | Arnsten AF , Rubia K . Neurobiological circuits regulating attention, cognitive control, motivation, and emotion: Disruptions in neurodevelopmental psychiatric disorders. J Am Acad Child Adolesc Psychiatry. (2012) ;51: (4):356–67. |
[62] | Barbas H . Connections underlying the synthesis of cognition, memory, and emotion in primate prefrontal cortices. Brain Res Bull. (2000) ;52: (5):319–30. |
[63] | Cummings JL . Anatomic and behavioral aspects of frontal-subcortical circuits. Ann N Y Acad Sci. (1995) ;769: :1–13. |
[64] | Gonzalez CE , Pacheco J , Beason-Held LL , Resnick SM . Longitudinal changes in cortical thinning associated with hypertension. J Hypertens. (2015) ;33: (6):1242–8. |
[65] | Gruner P , Pittenger C . Cognitive inflexibility in Obsessive-Compulsive Disorder. Neuroscience. (2017) ;345: :243–55. |
[66] | Murphy T , Dias GP , Thuret S . Effects of diet on brain plasticity in animal and human studies: Mind the gap. Neural Plast. (2014) ;2014: :563160. |
[67] | Voss MW , Vivar C , Kramer AF , van Praag H . Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn Sci. (2013) ;17: (10):525–44. |
[68] | Weintraub S , Carrillo MC , Farias ST , et al. Measuring cognition and function in the preclinical stage of Alzheimer’s disease. Alzheimers Dement (N Y). (2018) ;4: :64–75. |
[69] | Weintraub S , Wicklund AH , Salmon DP . The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med. (2012) ;2: (4):a006171. |
[70] | Salthouse TA . Shared and unique influences on age-related cognitive change. Neuropsychology. (2017) ;31: (1):11–9. |
[71] | McDonald AP , D’Arcy RCN , Song X . Functional MRI on executive functioning in aging and dementia: A scoping review of cognitive tasks. AGING MEDICINE. (2018) ;1: (2):209–19. |
[72] | Chow TW , Cummings JL . Frontal-Subcortical Circuits. In: Miller BL , Cummings JL , eds. The Human Frontal Lobes: Functions and Disorders. New York, London: Guilford Press; (2007) . |
[73] | Hsu CL , Best JR , Davis JC , et al. Aerobic exercise promotes executive functions and impacts functional neural activity among older adults with vascular cognitive impairment. Br J Sports Med. (2018) ;52: (3):184–91. |
[74] | da Silva FC , Iop RDR , de Oliveira LC , et al. Effects of physical exercise programs on cognitive function in Parkinson’s disease patients: A systematic review of randomized controlled trials of the last 10 years. PLoS One. (2018) ;13: (2):e0193113. |
[75] | Cerrillo-Urbina AJ , Garcia-Hermoso A , Sanchez-Lopez M , Pardo-Guijarro MJ , Santos Gomez JL , Martinez-Vizcaino V . The effects of physical exercise in children with attention deficit hyperactivity disorder: A systematic review and meta-analysis of randomized control trials. Child Care Health Dev. (2015) ;41: (6):779–88. |
[76] | Bischof GN , Park DC . Obesity and Aging: Consequences for Cognition, Brain Structure, and Brain Function. Psychosom Med. (2015) ;77: (6):697–709. |
[77] | Reuter-Lorenz PA , Park DC . How does it STAC up? Revisiting the scaffolding theory of aging and cognition. Neuropsychol Rev. (2014) ;24: (3):355–70. |
[78] | Park DC , McDonough IM . The Dynamic Aging Mind: Revelations From Functional Neuroimaging Research. Perspect Psychol Sci. (2013) ;8: (1):62–7. |
[79] | Stern Y . Elaborating a hypothetical concept: Comments on the special series on cognitive reserve. J Int Neuropsychol Soc. (2011) ;17: (4):639–42. |
[80] | Stern Y . Cognitive reserve. Neuropsychologia. (2009) ;47: (10):2015–28. |
[81] | Stern Y . Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord. (2006) ;20: (3 Suppl 2):S69–74. |
[82] | Gu Y , Brickman AM , Stern Y , et al. Mediterranean diet and brain structure in a multiethnic elderly cohort. Neurology. (2015) ;85: (20):1744–51. |
[83] | O’Hare C , Kenny RA , Aizenstein H , et al. Cognitive Status, Gray Matter Atrophy, and Lower Orthostatic Blood Pressure in Older Adults. J Alzheimers Dis. (2017) ;57: (4):1239–50. |
[84] | de la Torre JC . Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc Psychiatry Neurol. (2012) ;2012: :367516. |
[85] | Kennelly S , Collins O . Walking the cognitive “minefield” between high and low blood pressure. J Alzheimers Dis. (2012) ;32: (3):609–21. |
[86] | Stern Y , Gazes Y , Razlighi Q , Steffener J , Habeck C . A task-invariant cognitive reserve network. Neuroimage. (2018) ;178: :36–45. |
[87] | Bauckneht M , Chincarini A , Piva R , et al. Metabolic correlates of reserve and resilience in MCI due to Alzheimer’s Disease (AD). Alzheimers Res Ther. (2018) ;10: (1):35. |
[88] | Matura S , Fleckenstein J , Deichmann R , et al. Effects of aerobic exercise on brain metabolism and grey matter volume in older adults: Results of the randomised controlled SMART trial. Transl Psychiatry. (2017) ;7: (7):e1172. |
[89] | Stranahan AM , Mattson MP . Metabolic reserve as a determinant of cognitive aging. J Alzheimers Dis. (2012) ;30: (Suppl 2):S5–13. |
[90] | Perneczky R , Kempermann G , Korczyn AD , et al. Translational research on reserve against neurodegenerative disease: Consensus report of the International Conference on Cognitive Reserve in the Dementias and the Alzheimer’s Association Reserve, Resilience and Protective Factors Professional Interest Area working groups. BMC Med. (2019) ;17: (1):47. |
[91] | Spyridaki EC , Simos P , Avgoustinaki PD , et al. The association between obesity and fluid intelligence impairment is mediated by chronic low-grade inflammation. Br J Nutr. (2014) ;112: (10):1724–34. |
[92] | Guzzardi MA , Iozzo P . Brain functional imaging in obese and diabetic patients. Acta Diabetol. (2019) ;56: (2):135–44. |
[93] | Voss MW , Heo S , Prakash RS , et al. The influence of aerobic fitness on cerebral white matter integrity and cognitive function in older adults: Results of a one-year exercise intervention. Hum Brain Mapp. (2013) ;34: (11):2972–85. |
[94] | Oh H , Razlighi QR , Stern Y . Multiple pathways of reserve simultaneously present in cognitively normal older adults. Neurology. (2018) ;90: (3):e197–e205. |
[95] | Jackson PA , Pialoux V , Corbett D , et al. Promoting brain health through exercise and diet in older adults: A physiological perspective. J Physiol. (2016) ;594: (16):4485–98. |
[96] | Stern Y , Arenaza-Urquijo EM , Bartres-Faz D , et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement. (2018) . |
[97] | Kyriazis M . Nonlinear stimulation and hormesis in human aging: Practical examples and action mechanisms. Rejuvenation Res. (2010) ;13: (4):445–52. |
[98] | Gomez-Pinilla F . The influences of diet and exercise on mental health through hormesis. Ageing Res Rev. (2008) ;7: (1):49–62. |
[99] | Mapstone M , Cheema AK , Fiandaca MS , et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. (2014) ;20: (4):415–8. |
[100] | Wurtman R . Biomarkers in the diagnosis and management of Alzheimer’s disease. Metabolism. (2015) ;64: (3 Suppl 1):S47–50. |
[101] | Assi N , Gunter MJ , Thomas DC , et al. Metabolic signature of healthy lifestyle and its relation with risk of hepatocellular carcinoma in a large European cohort. Am J Clin Nutr. (2018) ;108: (1):117–26. |
[102] | Saleem M , Herrmann N , Dinoff A , et al. Association between sphingolipids and cardiopulmonary fitness in coronary artery disease patients undertaking cardiac rehabilitation. J Gerontol A Biol Sci Med Sci. (2018) . |
[103] | Kim M , Lee SH , Lee JH . Global Metabolic Profiling of Plasma Shows that Three-Year Mild-Caloric Restriction Lessens an Age-Related Increase in Sphingomyelin and Reduces L-leucine and L-phenylalanine in Overweight and Obese Subjects. Aging Dis. (2016) ;7: (6):721–33. |
[104] | Lacruz ME , Kluttig A , Tiller D , et al. Cardiovascular Risk Factors Associated With Blood Metabolite Concentrations and Their Alterations During a 4-Year Period in a Population-Based Cohort. Circ Cardiovasc Genet. (2016) ;9: (6):487–94. |
[105] | Bergman BC , Brozinick JT , Strauss A , et al. Serum sphingolipids: Relationships to insulin sensitivity and changes with exercise in humans. Am J Physiol Endocrinol Metab. (2015) ;309: (4):E398–408. |
[106] | Sumowski JF . “Brain reserve” and “cognitive reserve” should always be taken into account when studying neurodegeneration - YES. Mult Scler. (2018) ;24: (5):574–5. |
[107] | Daviglus ML , Bell CC , Berrettini W , et al. NIH State-of-the-Science Conference Statement: Preventing Alzheimer’s Disease and Cognitive Decline. NIH Consens State Sci Statements. (2010) ;27: (4). |
[108] | ADRD Summit 2016 Report to the National Advisory Neurological Disorders and Stroke Council. Alzheimer’s Disease-Related Dementias Summit 2016; 2016. |
[109] | Montine TJ , Koroshetz WJ , Babcock D , et al. Recommendations of the Alzheimer’s disease-related dementias conference. Neurology. (2014) ;83: (9):851–60. |
[110] | Hussenoeder FS , Riedel-Heller SG . Primary prevention of dementia: From modifiable risk factors to a public brain health agenda? Soc Psychiatry Psychiatr Epidemiol. (2018) ;53: (12):1289–301. |
[111] | Hersi M , Irvine B , Gupta P , Gomes J , Birkett N , Krewski D . Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology. (2017) ;61: :143–87. |
[112] | Sofi F , Valecchi D , Bacci D , et al. Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. Journal of Internal Medicine. (2011) ;269: (1):107–17. |
[113] | Radd-Vagenas S , Duffy SL , Naismith SL , Brew BJ , Flood VM , Fiatarone Singh MA . Effect of the Mediterranean diet on cognition and brain morphology and function: A systematic review of randomized controlled trials. Am J Clin Nutr. (2018) ;107: (3):389–404. |
[114] | Wu L , Sun D , Tan Y . Intake of Fruit and Vegetables and the Incident Risk of Cognitive Disorders: A Systematic Review and Meta-Analysis of Cohort Studies. J Nutr Health Aging. (2017) ;21: (10):1284–90. |
[115] | Aarsland D , Sardahaee FS , Anderssen S , Ballard C . Is physical activity a potential preventive factor for vascular dementia? A systematic review. Aging Ment Health. (2010) ;14: (4):386–95. |
[116] | Angevaren M , Aufdemkampe G , Verhaar HJ , Aleman A , Vanhees L . Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. CochraneDatabaseSystRev. 2008: (3):CD005381. |
[117] | Smith PJ , Blumenthal JA , Hoffman BM , et al. Aerobic exercise and neurocognitive performance: A meta-analytic review of randomized controlled trials. Psychosom Med. (2010) ;72: (3):239–52. |
[118] | Colcombe S , Kramer AF . Fitness effects on the cognitive function of older adults: A meta-analytic study. Psychol Sci. (2003) ;14: (2):125–30. |
[119] | Wang S , Luo X , Barnes D , Sano M , Yaffe K . Physical activity and risk of cognitive impairment among oldest-old women. Am J Geriatr Psychiatry. (2014) ;22: (11):1149–57. |
[120] | Lee Y , Back JH , Kim J , et al. Systematic review of health behavioral risks and cognitive health in older adults. Int Psychogeriatr. (2010) ;22: (2):174–87. |
[121] | Sabia S , Dugravot A , Dartigues JF , et al. Physical activity, cognitive decline, and risk of dementia: 28 year follow-up of Whitehall II cohort study. BMJ. (2017) ;357: :j2709. |
[122] | Guure CB , Ibrahim NA , Adam MB , Said SM . Impact of Physical Activity on Cognitive Decline, Dementia, and Its Subtypes: Meta-Analysis of Prospective Studies. Biomed Res Int. (2017) ;2017: :9016924. |
[123] | Jensen CS , Simonsen AH , Siersma V , et al. Patients with Alzheimer’s disease who carry the APOE epsilon4 allele benefit more from physical exercise. Alzheimers Dement (N Y). (2019) ;5: :99–106. |
[124] | Berkowitz CL , Mosconi L , Rahman A , Scheyer O , Hristov H , Isaacson RS . Clinical Application of APOE in Alzheimer’s Prevention: A Precision Medicine Approach. The Journal of Prevention of Alzheimer’s Disease. (2018) ;5: (4):245–52. |
[125] | Etnier JL , Karper WB , Labban JD , et al. The Physical Activity and Alzheimer’s Disease (PAAD) Study: Cognitive outcomes. Ann Behav Med. (2018) ;52: (2):175–85. |
[126] | Solomon A , Turunen H , Ngandu T , et al. Effect of the Apolipoprotein E Genotype on Cognitive Change During a Multidomain Lifestyle Intervention: A Subgroup Analysis of a Randomized Clinical Trial. JAMA Neurology. (2018) ;75: (4):462–70. |
[127] | Liang JH , Xu Y , Lin L , Jia RX , Zhang HB , Hang L . Comparison of multiple interventions for older adults with Alzheimer disease or mild cognitive impairment: A PRISMA-compliant network meta-analysis. Medicine (Baltimore). (2018) ;97: (20):e10744. |
[128] | Kelly ME , Loughrey D , Lawlor BA , Robertson IH , Walsh C , Brennan S . The impact of exercise on the cognitive functioning of healthy older adults: A systematic review and meta-analysis. Ageing Res Rev. (2014) ;16: :12–31. |
[129] | Northey JM , Cherbuin N , Pumpa KL , Smee DJ , Rattray B . Exercise interventions for cognitive function in adults older than 50: A systematic review with meta-analysis. Br J Sports Med. (2017) . |
[130] | Mavros Y , Gates N , Wilson GC , et al. Mediation of Cognitive Function Improvements by Strength Gains After Resistance Training in Older Adults with Mild Cognitive Impairment: Outcomes of the Study of Mental and Resistance Training. Journal of the American Geriatrics Society. (2017) ;65: (3):550–9. |
[131] | Li Z , Peng X , Xiang W , Han J , Li K . The effect of resistance training on cognitive function in the older adults: A systematic review of randomized clinical trials. Aging Clin Exp Res. (2018) ;30: (11):1259–73. |
[132] | Yoon DH , Lee JY , Song W . Effects of Resistance Exercise Training on Cognitive Function and Physical Performance in Cognitive Frailty: A Randomized Controlled Trial. J Nutr Health Aging. (2018) ;22: (8):944–51. |
[133] | Sanders LMJ , Hortobagyi T , la Bastide-van Gemert S , van der Zee EA , van Heuvelen MJG . Dose-response relationship between exercise and cognitive function in older adults with and without cognitive impairment: A systematic review and meta-analysis. PLoS One. (2019) ;14: (1):e0210036. |
[134] | Dascal JB , Sanders LMJ , Filho E , Hortobagyi T . Dose-response effects of years of self-reported physical activity on old females’ motor and cognitive function. Braz J Phys Ther. (2019) ;23: (1):48–55. |
[135] | Loprinzi PD , Edwards MK , Crush E , Ikuta T , Del Arco A . Dose-Response Association Between Physical Activity and Cognitive Function in a National Sample of Older Adults. Am J Health Promot. (2018) ;32: (3):554–60. |
[136] | Etnier JL , Nowell PM , Landers DM , Sibley BA . A meta-regression to examine the relationship between aerobic fitness and cognitive performance. Brain Res Brain Res Rev. (2006) ;52: (1):119–30. |
[137] | Anastasiou CA , Yannakoulia M , Kosmidis MH , et al. Mediterranean diet and cognitive health: Initial results from the Hellenic Longitudinal Investigation of Ageing and Diet. PLoS One. (2017) ;12: (8):e0182048. |
[138] | Daviglus ML , Plassman BL , Pirzada A , et al. Risk factors and preventive interventions for Alzheimer disease: State of the science. Arch Neurol. (2011) ;68: (9):1185–90. |
[139] | Demarin V , Lisak M , Morovic S . Mediterranean diet in healthy lifestyle and prevention of stroke. Acta Clin Croat. (2011) ;50: (1):67–77. |
[140] | Feart C , Samieri C , Barberger-Gateau P . Mediterranean diet and cognitive function in older adults. CurrOpinClinNutrMetab Care. (2010) ;13: (1):14–8. |
[141] | Feart C , Samieri C , Barberger-Gateau P . Mediterranean diet and cognitive health: An update of available knowledge. Curr Opin Clin Nutr Metab Care. (2015) ;18: (1):51–62. |
[142] | Blumenthal JA , Smith PJ , Mabe S , et al. Lifestyle and Neurocognition in Older Adults with Cardiovascular Risk Factors and Cognitive Impairment. Psychosomatic medicine. (2017) . |
[143] | Blumenthal JA , Smith PJ , Welsh-Bohmer K , et al. Can lifestyle modification improve neurocognition? Rationale and design of the ENLIGHTEN clinical trial. Contemp Clin Trials. (2013) ;34: (1):60–9. |
[144] | Smith PJ , Blumenthal JA . Dietary Factors and Cognitive Decline. The Journal of Prevention of Alzheimer’s Disease. (2016) ;3: (1):53–64. |
[145] | Smith PJ , Blumenthal JA , Babyak MA , et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. (2010) ;55: (6):1331–8. |
[146] | Tangney CC , Li H , Wang Y , et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. (2014) ;83: (16):1410–6. |
[147] | Wengreen H , Munger RG , Cutler A , et al. Prospective study of Dietary Approaches to Stop Hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: The Cache County Study on Memory, Health and Aging. Am J Clin Nutr. (2013) ;98: (5):1263–71. |
[148] | Abbatecola AM , Russo M , Barbieri M . Dietary patterns and cognition in older persons. Curr Opin Clin Nutr Metab Care. (2018) ;21: (1):10–3. |
[149] | Morris MC . Nutrition and risk of dementia: Overview and methodological issues. Ann N Y Acad Sci. (2016) ;1367: (1):31–7. |
[150] | Morris MC , Tangney CC , Wang Y , et al. MIND diet slows cognitive decline with aging. Alzheimers Dement. (2015) ;11: (9):1015–22. |
[151] | Knight A , Bryan J , Murphy K . The Mediterranean diet and age-related cognitive functioning: A systematic review of study findings and neuropsychological assessment methodology. Nutr Neurosci. (2016) :1–20. |
[152] | Knight A , Bryan J , Murphy K . Is the Mediterranean diet a feasible approach to preserving cognitive function and reducing risk of dementia for older adults in Western countries? New insights and future directions. Ageing Res Rev. (2016) ;25: :85–101. |
[153] | Scarmeas N , Stern Y , Mayeux R , Luchsinger JA . Mediterranean diet, Alzheimer disease, and vascular mediation. Arch Neurol. (2006) ;63: (12):1709–17. |
[154] | Smith PJ , Blumenthal JA . Diet and neurocognition: Review of evidence and methodological considerations. Curr Aging Sci. (2010) ;3: (1):57–66. |
[155] | Scarmeas N , Stern Y , Tang MX , Mayeux R , Luchsinger JA . Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. (2006) ;59: (6):912–21. |
[156] | Solfrizzi V , Custodero C , Lozupone M , et al. Relationships of Dietary Patterns, Foods, and Micro- and Macronutrients with Alzheimer’s Disease and Late-Life Cognitive Disorders: A Systematic Review. J Alzheimers Dis. (2017) ;59: (3):815–49. |
[157] | Cao L , Tan L , Wang HF , et al. Dietary Patterns and Risk of Dementia: A Systematic Review and Meta-Analysis of Cohort Studies. Mol Neurobiol. (2016) ;53: (9):6144–54. |
[158] | Singh B , Parsaik AK , Mielke MM , et al. Association of mediterranean diet with mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis. J Alzheimers Dis. (2014) ;39: (2):271–82. |
[159] | Mosconi L , Murray J , Tsui WH , Li Y , Davies M , Williams S , Pirraglia E , Spector N , Osorio RS , Glodzik L , McHugh P , de Leon MJ . Mediterranean Diet and Magnetic Resonance Imaging-Assessed Brain Atrophy in Cognitively Normal Individuals at Risk for Alzheimer’s Disease. J Prev Alzheimers Dis. (2014) ;1: (1):23–32. |
[160] | Estruch R , Ros E , Salas-Salvado J , et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med. (2013) ;368: (14):1279–90. |
[161] | Martinez-Lapiscina EH , Clavero P , Toledo E , et al. Mediterranean diet improves cognition: The PREDIMED-NAVARRA randomised trial. J Neurol Neurosurg Psychiatry. (2013) . |
[162] | Wu L , Sun D . Adherence to Mediterranean diet and risk of developing cognitive disorders: An updated systematic review and meta-analysis of prospective cohort studies. Sci Rep. (2017) ;7: :41317. |
[163] | Epstein DE , Sherwood A , Smith PJ , et al. Determinants and consequences of adherence to the dietary approaches to stop hypertension diet in African-American and white adults with high blood pressure: Results from the ENCORE trial. J Acad Nutr Diet. (2012) ;112: (11):1763–73. |
[164] | Fung TT , Chiuve SE , McCullough ML , Rexrode KM , Logroscino G , Hu FB . Adherence to a DASH-style diet and risk of coronary heart disease and stroke in women. Arch Intern Med. (2008) ;168: (7):713–20. |
[165] | Chen X , Maguire B , Brodaty H , O’Leary F . Dietary Patterns and Cognitive Health in Older Adults: A Systematic Review. J Alzheimers Dis. (2019) ;67: (2):583–619. |
[166] | van den Brink AC , Brouwer-Brolsma EM , Berendsen AAM , van de Rest O . The Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) Diets Are Associated with Less Cognitive Decline and a Lower Risk of Alzheimer’s Disease-A Review. Adv Nutr. (2019) . |
[167] | Hosking DE , Eramudugolla R , Cherbuin N , Anstey KJ . MIND not Mediterranean diet related to 12-year incidence of cognitive impairment in an Australian longitudinal cohort study. Alzheimers Dement. (2019) ;15: (4):581–9. |
[168] | Berendsen AM , Kang JH , Feskens EJM , de Groot C , Grodstein F , van de Rest O . Association of Long-Term Adherence to the MIND Diet with Cognitive Function and Cognitive Decline in American Women. J Nutr Health Aging. (2018) ;22: (2):222–9. |
[169] | Omar SH . Mediterranean and MIND Diets Containing Olive Biophenols Reduces the Prevalence of Alzheimer’s Disease. Int J Mol Sci. (2019) ;20: (11). |
[170] | Feart C , Samieri C , Rondeau V , et al. Adherence to a Mediterranean diet, cognitive decline, and risk of dementia. JAMA: The Journal of the American Medical Association. (2009) ;302: (6):638–48. |
[171] | Koch M , Jensen MK . Association of the MIND diet with cognition and risk of Alzheimer’s disease. Curr Opin Lipidol. (2016) ;27: (3):303–4. |
[172] | Morris MC , Tangney CC , Wang Y , Sacks FM , Bennett DA , Aggarwal NT . MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. (2015) ;11: (9):1007–14. |
[173] | Saito S , Yamamoto Y , Ihara M . Development of a Multicomponent Intervention to Prevent Alzheimer’s Disease. Front Neurol. (2019) ;10: :490. |
[174] | Gillette-Guyonnet S , Vellas B . Caloric restriction and brain function. CurrOpinClinNutrMetab Care. (2008) ;11: (6):686–92. |
[175] | Luchsinger JA , Noble JM , Scarmeas N . Diet and Alzheimer’s disease. CurrNeurolNeurosciRep. (2007) ;7: (5):366–72. |
[176] | Siervo M , Arnold R , Wells JC , et al. Intentional weight loss in overweight and obese individuals and cognitive function: A systematic review and meta-analysis. Obes Rev. (2011) ;12: (11):968–83. |
[177] | Willcox BJ , Willcox DC , Todoriki H , et al. Caloric restriction, the traditional Okinawan diet, and healthy aging: The diet of the world’s longest-lived people and its potential impact on morbidity and life span. Ann N Y AcadSci. (2007) ;1114: :434–55. |
[178] | Witte AV , Fobker M , Gellner R , Knecht S , Floel A . Caloric restriction improves memory in elderly humans. ProcNatlAcadSci US A. (2009) ;106: (4):1255–60. |
[179] | Luchsinger JA , Tang MX , Shea S , Mayeux R . Caloric intake and the risk of Alzheimer disease. Arch Neurol. (2002) ;59: (8):1258–63. |
[180] | Martin CK , Anton SD , Han H , et al. Examination of cognitive function during six months of calorie restriction: Results of a randomized controlled trial. Rejuvenation Res. (2007) ;10: (2):179–90. |
[181] | Bryan J , Tiggemann M . The effect of weight-loss dieting on cognitive performance and psychological well-being in overweight women. Appetite. (2001) ;36: (2):147–56. |
[182] | Halyburton AK , Brinkworth GD , Wilson CJ , et al. Low- and high-carbohydrate weight-loss diets have similar effects on mood but not cognitive performance. Am J Clin Nutr. (2007) ;86: (3):580–7. |
[183] | Rochon J , Bales CW , Ravussin E , et al. Design and conduct of the CALERIE study: Comprehensive assessment of the long-term effects of reducing intake of energy. The journals of gerontology Series A, Biological sciences and medical sciences. (2011) ;66: (1):97–108. |
[184] | Leclerc E , Trevizol AP , Grigolon RB , et al. The effect of caloric restriction on working memory in healthy non-obese adults. CNS Spectr. (2019) :1–7. |
[185] | Trevizol AP , Brietzke E , Grigolon RB , Subramaniapillai M , McIntyre RS , Mansur RB . Peripheral interleukin-6 levels and working memory in non-obese adults: A post-hoc analysis from the CALERIE study. Nutrition. (2019) ;58: :18–22. |
[186] | Horie NC , Serrao VT , Simon SS , et al. Cognitive Effects of Intentional Weight Loss in Elderly Obese Individuals With Mild Cognitive Impairment. J Clin Endocrinol Metab. (2016) ;101: (3):1104–12. |
[187] | Prehn K , Jumpertz von Schwartzenberg R , Mai K , et al. Caloric Restriction in Older Adults-Differential Effects of Weight Loss and Reduced Weight on Brain Structure and Function. Cerebral cortex. (2016) . |
[188] | Harvie MN , Pegington M , Mattson MP , et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: A randomized trial in young overweight women. Int J Obes (Lond). (2011) ;35: (5):714–27. |
[189] | Martin B , Mattson MP , Maudsley S . Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Res Rev. (2006) ;5: (3):332–53. |
[190] | Cherif A , Roelands B , Meeusen R , Chamari K . Effects of Intermittent Fasting, Caloric Restriction, and Ramadan Intermittent Fasting on Cognitive Performance at Rest and During Exercise in Adults. Sports Med. (2016) ;46: (1):35–47. |
[191] | Davis CS , Clarke RE , Coulter SN , et al. Intermittent energy restriction and weight loss: A systematic review. Eur J Clin Nutr. (2016) ;70: (3):292–9. |
[192] | Klempel MC , Kroeger CM , Bhutani S , Trepanowski JF , Varady KA . Intermittent fasting combined with calorie restriction is effective for weight loss and cardio-protection in obese women. Nutrition Journal. (2012) ;11: :98. |
[193] | Kroeger CM , Klempel MC , Bhutani S , Trepanowski JF , Tangney CC , Varady KA . Improvement in coronary heart disease risk factors during an intermittent fasting/calorie restriction regimen: Relationship to adipokine modulations. Nutrition & Metabolism. (2012) ;9: (1):98. |
[194] | Halagappa VK , Guo Z , Pearson M , et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. (2007) ;26: (1):212–20. |
[195] | Varady KA . Intermittent versus daily calorie restriction: Which diet regimen is more effective for weight loss? Obes Rev. (2011) ;12: (7):e593–601. |
[196] | Verrotti A , Iapadre G , Pisano S , Coppola G . Ketogenic diet and childhood neurological disorders other than epilepsy: An overview. Expert Rev Neurother. (2017) ;17: (5):461–73. |
[197] | Cunnane SC , Courchesne-Loyer A , St-Pierre V , et al. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann N Y Acad Sci. (2016) ;1367: (1):12–20. |
[198] | Hertz L , Chen Y , Waagepetersen HS . Effects of ketone bodies in Alzheimer’s disease in relation to neural hypometabolism, beta-amyloid toxicity, and astrocyte function. J Neurochem. (2015) ;134: (1):7–20. |
[199] | Iacovides S , Goble D , Paterson B , Meiring RM . Three consecutive weeks of nutritional ketosis has no effect on cognitive function, sleep, and mood compared with a high-carbohydrate, low-fat diet in healthy individuals: A randomized, crossover, controlled trial. Am J Clin Nutr. (2019) . |
[200] | Mohorko N , Cernelic-Bizjak M , Poklar-Vatovec T , et al. Weight loss, improved physical performance, cognitive function, eating behavior, and metabolic profile in a 12-week ketogenic diet in obese adults. Nutr Res. (2019) ;62: :64–77. |
[201] | van Berkel AA , DM IJ , Verkuyl JM . Cognitive benefits of the ketogenic diet in patients with epilepsy: A systematic overview. Epilepsy Behav. (2018) ;87: :69–77. |
[202] | Hernandez AR , Hernandez CM , Campos K , et al. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable From Hippocampus. Front Aging Neurosci. (2018) ;10: :391. |
[203] | Blumenthal JA , Smith PJ , Welsh-Bohmer K , et al. Can lifestyle modification improve neurocognition? Rationale and design of the ENLIGHTEN clinical trial. Contemp Clin Trials. (2013) ;34: (1):60–9. |
[204] | Blumenthal JA , Smith PJ , Mabe S , et al. Lifestyle and Neurocognition in Older Adults With Cardiovascular Risk Factors and Cognitive Impairment. Psychosom Med. (2017) ;79: (6):719–27. |
[205] | Zhao C , Noble JM , Marder K , Hartman JS , Gu Y , Scarmeas N . Dietary Patterns, Physical Activity, Sleep, and Risk for Dementia and Cognitive Decline. Curr Nutr Rep. (2018) ;7: (4):335–45. |
[206] | Scarmeas N , Luchsinger JA , Schupf N , et al. Physical Activity, Diet, and Risk of Alzheimer Disease. JAMA: The Journal of the American Medical Association. (2009) ;302: (6):627–37. |
[207] | Intlekofer KA , Berchtold NC , Malvaez M , et al. Exercise and sodium butyrate transform a subthreshold learning event into long-term memory via a brain-derived neurotrophic factor-dependent mechanism. Neuropsychopharmacology. (2013) ;38: (10):2027–34. |
[208] | Head E , Nukala VN , Fenoglio KA , Muggenburg BA , Cotman CW , Sullivan PG . Effects of age, dietary, and behavioral enrichment on brain mitochondria in a canine model of human aging. Exp Neurol. (2009) ;220: (1):171–6. |
[209] | Wu A , Ying Z , Gomez-Pinilla F . Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience. (2013) ;248: :655–63. |
[210] | Gomez-Pinilla F , Gomez AG . The influence of dietary factors in central nervous system plasticity and injury recovery. Pm R. (2011) ;3: (6 Suppl 1):S111–116. |
[211] | Gomez-Pinilla F . The combined effects of exercise and foods in preventing neurological and cognitive disorders. Prev Med. (2011) ;52: (Suppl 1):S75–80. |
[212] | Griesbach GS , Hovda DA , Gomez-Pinilla F . Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. (2009) ;1288: :105–15. |
[213] | Marengoni A , Rizzuto D , Fratiglioni L , et al. The Effect of a 2-Year Intervention Consisting of Diet, Physical Exercise, Cognitive Training, and Monitoring of Vascular Risk on Chronic Morbidity-the FINGER Randomized Controlled Trial. J Am Med Dir Assoc. (2018) ;19: (4):355–360. e351 |
[214] | Ngandu T , Lehtisalo J , Solomon A , et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet. (2015) ;385: (9984):2255–63. |
[215] | Espeland MA , Carmichael O , Hayden K , et al. Long-term Impact of Weight Loss Intervention on Changes in Cognitive Function: Exploratory Analyses from the Action for Health in Diabetes Randomized Controlled Clinical Trial. The journals of gerontology Series A, Biological Sciences and Medical Sciences. (2018) ;73: (4):484–91. |
[216] | Espeland MA , Lipska K , Miller ME , et al. Effects of Physical Activity Intervention on Physical and Cognitive Function in Sedentary Adults With and Without Diabetes. J Gerontol A Biol Sci Med Sci. (2017) ;72: (6):861–6. |
[217] | Espeland MA , Luchsinger JA , Baker LD , et al. Effect of a long-term intensive lifestyle intervention on prevalence of cognitive impairment. Neurology. (2017) ;88: (21):2026–35. |
[218] | Rapp SR , Luchsinger JA , Baker LD , et al. Effect of a Long-Term Intensive Lifestyle Intervention on Cognitive Function: Action for Health in Diabetes Study. J Am Geriatr Soc. (2017) ;65: (5):966–72. |
[219] | Sink KM , Espeland MA , Castro CM , et al. Effect of a 24-Month Physical Activity Intervention vs Health Education on Cognitive Outcomes in Sedentary Older Adults: The LIFE Randomized Trial. Jama. (2015) ;314: (8):781–90. |
[220] | Hayden KM , Baker LD , Bray G , et al. Long-term impact of intensive lifestyle intervention on cognitive function assessed with the National Institutes of Health Toolbox: The Look AHEAD study. Alzheimers Dement (Amst). (2018) ;10: :41–8. |
[221] | Espeland MA , Erickson K , Neiberg RH , et al. Brain and White Matter Hyperintensity Volumes After 10 Years of Random Assignment to Lifestyle Intervention. Diabetes Care. (2016) ;39: (5):764–71. |
[222] | Casanova R , Hayasaka S , Saldana S , et al. Relative differences in resting-state brain connectivity associated with long term intensive lifestyle intervention. Psychoneuroendocrinology. (2016) ;74: :231–9. |
[223] | Kivipelto M , Solomon A , Ahtiluoto S , et al. The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER): Study design and progress. Alzheimers Dement. (2013) ;9: (6):657–65. |
[224] | Stephen R , Liu Y , Ngandu T , et al. Brain volumes and cortical thickness on MRI in the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). Alzheimers Res Ther. (2019) ;11: (1):53. |
[225] | Pentikainen H , Savonen K , Ngandu T , et al. Cardiorespiratory Fitness and Cognition: Longitudinal Associations in the FINGER Study. J Alzheimers Dis. (2019) ;68: (3):961–8. |
[226] | Lehtisalo J , Levalahti E , Lindstrom J , et al. Dietary changes and cognition over 2 years within a multidomain intervention trial-The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). Alzheimers Dement. (2019) ;15: (3):410–7. |
[227] | Stephen R , Liu Y , Ngandu T , et al. Associations of CAIDE Dementia Risk Score with MRI, PIB-PET measures, and cognition. J Alzheimers Dis. (2017) ;59: (2):695–705. |
[228] | Moll van Charante EP , Richard E , Eurelings LS , et al. Effectiveness of a 6-year multidomain vascular care intervention to prevent dementia (preDIVA): A cluster-randomised controlled trial. Lancet. (2016) ;388: (10046):797–805. |
[229] | Kivipelto M , Mangialasche F , Ngandu T . Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol. (2018) ;14: (11):653–66. |
[230] | Moll van Charante EP , Richard E , Eurelings LS , et al. [Is dementia preventable through intensive vascular care? The preDIVA trial]. Ned Tijdschr Geneeskd. (2017) ;161: :D1184. |
[231] | Andrieu S , Guyonnet S , Coley N , et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): A randomised, placebo-controlled trial. Lancet Neurol. (2017) ;16: (5):377–89. |
[232] | Vellas B , Carrie I , Gillette-Guyonnet S , et al. MAPT STUDY: A MULTIDOMAIN APPROACH FOR PREVENTING ALZHEIMER’S DISEASE: DESIGN AND BASELINE DATA. The journal of prevention of Alzheimer’s disease. (2014) ;1: (1):13–22. |
[233] | Koekkoek PS , Ruis C , van den Donk M , et al. Intensive multifactorial treatment and cognitive functioning in screen-detected type 2 diabetes–the ADDITION-Netherlands study: A cluster-randomized trial. J Neurol Sci. (2012) ;314: (1-2):71–7. |
[234] | Simmons RK , Echouffo-Tcheugui JB , Sharp SJ , et al. Screening for type 2 diabetes and population mortality over 10 years (ADDITION-Cambridge): A cluster-randomised controlled trial. Lancet. (2012) ;380: (9855):1741–8. |
[235] | Griffin SJ , Borch-Johnsen K , Davies MJ , et al. Effect of early intensive multifactorial therapy on 5-year cardiovascular outcomes in individuals with type 2 diabetes detected by screening (ADDITION-Europe): A cluster-randomised trial. Lancet. (2011) ;378: (9786):156–67. |
[236] | Blumenthal JA , Smith PJ , Mabe S , et al. Lifestyle and neurocognition in older adults with cognitive impairments: A randomized trial. Neurology. (2019) ;92: (3):e212–e223. |
[237] | Blumenthal JA , Babyak MA , Sherwood A , et al. Effects of the dietary approaches to stop hypertension diet alone and in combination with exercise and caloric restriction on insulin sensitivity and lipids. Hypertension. (2010) ;55: (5):1199–205. |
[238] | Blumenthal JA , Babyak MA , Hinderliter A , et al. Effects of the DASH diet alone and in combination with exercise and weight loss on blood pressure and cardiovascular biomarkers in men and women with high blood pressure: The ENCORE study. Arch Intern Med. (2010) ;170: (2):126–35. |
[239] | Martinez-Lapiscina EH , Clavero P , Toledo E , et al. Mediterranean diet improves cognition: The PREDIMED-NAVARRA randomised trial. J Neurol Neurosurg Psychiatry. (2013) ;84: (12):1318–25. |
[240] | Valls-Pedret C , Sala-Vila A , Serra-Mir M , et al. Mediterranean Diet and Age-Related Cognitive Decline: A Randomized Clinical Trial. JAMA Internal Medicine. (2015) ;175: (7):1094–103. |
[241] | Martinez-Lapiscina EH , Clavero P , Toledo E , et al. Virgin olive oil supplementation and long-term cognition: The PREDIMED-NAVARRA randomized, trial. J Nutr Health Aging. (2013) ;17: (6):544–52. |
[242] | Espeland MA , Rapp SR , Bray GA , et al. Long-term impact of behavioral weight loss intervention on cognitive function. The journals of gerontology Series A, Biological Sciences and Medical Sciences. (2014) ;69: (9):1101–8. |
[243] | Look ARG , Wadden TA , West DS , et al. The Look AHEAD study: A description of the lifestyle intervention and the evidence supporting it. Obesity (Silver Spring). (2006) ;14: (5):737–52. |
[244] | Espeland MA , Luchsinger JA , Neiberg RH , et al. Long Term Effect of Intensive Lifestyle Intervention on Cerebral Blood Flow. J Am Geriatr Soc. (2018) ;66: (1):120–6. |
[245] | Napoli N , Shah K , Waters DL , Sinacore DR , Qualls C , Villareal DT . Effect of weight loss, exercise, or both on cognition and quality of life in obese older adults. Am J Clin Nutr. (2014) ;100: (1):189–98. |
[246] | Cotman CW , Berchtold NC . Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. (2002) ;25: (6):295–301. |
[247] | Cotman CW , Berchtold NC . Physical activity and the maintenance of cognition: Learning from animal models. Alzheimers Dement. (2007) ;3: (2 Suppl):S30–37. |
[248] | Hardman RJ , Kennedy G , Macpherson H , Scholey AB , Pipingas A . A randomised controlled trial investigating the effects of Mediterranean diet and aerobic exercise on cognition in cognitively healthy older people living independently within aged care facilities: The Lifestyle Intervention in Independent Living Aged Care (LIILAC) study protocol [ACTRN12614001133628]. Nutr J. (2015) ;14: :53. |
[249] | Woo J , Shin KO , Park SY , Jang KS , Kang S . Effects of exercise and diet change on cognition function and synaptic plasticity in high fat diet induced obese rats. Lipids Health Dis. (2013) ;12: :144. |
[250] | Foster PP , Rosenblatt KP , Kuljis RO . Exercise-induced cognitive plasticity, implications for mild cognitive impairment and Alzheimer’s disease. Front Neurol. (2011) ;2: :28. |
[251] | Maass A , Duzel S , Brigadski T , et al. Relationships of peripheral IGF-1, VEGF and BDNF levels to exercise-related changes in memory, hippocampal perfusion and volumes in older adults. Neuroimage. (2016) ;131: :142–54. |
[252] | Pakiz B , Flatt SW , Bardwell WA , Rock CL , Mills PJ . Effects of a weight loss intervention on body mass, fitness, and inflammatory biomarkers in overweight or obese breast cancer survivors. Int J Behav Med. (2011) ;18: (4):333–41. |
[253] | Tumminia A , Vinciguerra F , Parisi M , Frittitta L . Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int J Mol Sci. (2018) ;19: (11). |
[254] | Culberson JW . Clinical Aspects of Glucose Metabolism and Chronic Disease. Prog Mol Biol Transl Sci. (2017) ;146: :1–11. |
[255] | Rani V , Deshmukh R , Jaswal P , Kumar P , Bariwal J . Alzheimer’s disease: Is this a brain specific diabetic condition? Physiol Behav. (2016) ;164: (Pt A):259–67. |
[256] | Leckie RL , Oberlin LE , Voss MW , et al. BDNF mediates improvements in executive function following a 1-year exercise intervention. Frontiers in Human Neuroscience. (2014) ;8: :985. |
[257] | Vaughan S , Wallis M , Polit D , Steele M , Shum D , Morris N . The effects of multimodal exercise on cognitive and physical functioning and brain-derived neurotrophic factor in older women: A randomised controlled trial. Age and Ageing. (2014) ;43: (5):623–9. |
[258] | Marosi K , Mattson MP . BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol Metab. (2014) ;25: (2):89–98. |
[259] | Vivar C , Potter MC , van Praag H . All About Running: Synaptic Plasticity, Growth Factors and Adult Hippocampal Neurogenesis. Curr Top Behav Neurosci. (2012) . |
[260] | Erickson KI , Voss MW , Prakash RS , et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. (2011) ;108: (7):3017–22. |
[261] | Griffin EW , Bechara RG , Birch AM , Kelly AM . Exercise enhances hippocampal-dependent learning in the rat: Evidence for a BDNF-related mechanism. Hippocampus. (2009) ;19: (10):973–80. |
[262] | Rothman SM , Griffioen KJ , Wan R , Mattson MP . Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Ann N Y Acad Sci. (2012) ;1264: (1):49–63. |
[263] | Hohman TJ , Bell SP , Jefferson AL , Alzheimer’s Disease Neuroimaging I. The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: Exploring interactions with biomarkers of Alzheimer disease. JAMA Neurology. (2015) ;72: (5):520–9. |
[264] | Avolio A , Kim MO , Adji A , et al. Cerebral Haemodynamics: Effects of Systemic Arterial Pulsatile Function and Hypertension. Curr Hypertens Rep. (2018) ;20: (3):20. |
[265] | Toth P , Tarantini S , Csiszar A , Ungvari Z . Functional vascular contributions to cognitive impairment and dementia: Mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. (2017) ;312: (1):H1–H20. |
[266] | Grammas P . Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation. (2011) ;8: :26. |
[267] | Smith PJ , Blumenthal JA , Babyak MA , et al. Cerebrovascular risk factors, vascular disease, and neuropsychological outcomes in adults with major depression. Psychosomatic medicine. (2007) ;69: (6):578–86. |
[268] | Smith PJ , Blumenthal JA , Babyak MA , Hinderliter A , Sherwood A . Association of vascular health and neurocognitive performance in overweight adults with high blood pressure. J Clin Exp Neuropsychol. (2011) ;33: (5):559–66. |
[269] | Najjar S , Pahlajani S , De Sanctis V , Stern JNH , Najjar A , Chong D . Neurovascular Unit Dysfunction and Blood-Brain Barrier Hyperpermeability Contribute to Schizophrenia Neurobiology: A Theoretical Integration of Clinical and Experimental Evidence. Front Psychiatry. (2017) ;8: :83. |
[270] | Nindl BC , Pierce JR , Insulin-like growth factor I as a biomarker of health, fitness, and training status. Med Sci Sports Exerc. (2010) ;42: (1):39–49. |
[271] | Anderson JR , Calvo D , Glickman E , Gunstad J , Spitznagel MB . The Moderating Role of Insulin-Like Growth Factor 1 in the Relationship Between Cognitive and Aerobic Endurance Change. J Geriatr Psychiatry Neurol. (2017) ;30: (2):84–9. |
[272] | Spielman LJ , Little JP , Klegeris A . Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J Neuroimmunol. (2014) ;273: (1-2):8–21. |
[273] | de la Monte SM . Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. (2012) ;9: (1):35–66. |
[274] | de la Monte SM , Tong M . Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol. (2014) ;88: (4):548–59. |
[275] | Stein AM , Silva TMV , Coelho FGM , et al. Physical exercise, IGF-1 and cognition A systematic review of experimental studies in the elderly. Dement Neuropsychol. (2018) ;12: (2):114–22. |
[276] | Tsai CL , Pai MC , Ukropec J , Ukropcova B . Distinctive Effects of Aerobic and Resistance Exercise Modes on Neurocognitive and Biochemical Changes in Individuals with Mild Cognitive Impairment. Curr Alzheimer Res. (2019) ;16: (4):316–32. |
[277] | McGrattan AM , McGuinness B , McKinley MC , et al. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr Nutr Rep. (2019) ;8: (2):53–65. |
[278] | Van Dyken P , Lacoste B . Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front Neurosci. (2018) ;12: :930. |
[279] | Najem D , Bamji-Mirza M , Chang N , Liu QY , Zhang W . Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev Neurosci. (2014) ;25: (4):509–25. |
[280] | Ferreira ST , Clarke JR , Bomfim TR , De Felice FG . Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. (2014) ;10: (1 Suppl):S76–83. |
[281] | Vos RC , Wit JM , Pijl H , Houdijk EC . Long-term effect of lifestyle intervention on adiposity, metabolic parameters, inflammation and physical fitness in obese children: A randomized controlled trial. Nutr Diabetes. (2011) ;1: :e9. |
[282] | Hoth KF , Haley AP , Gunstad J , et al. Elevated C-reactive protein is related to cognitive decline in older adults with cardiovascular disease. Journal of the American Geriatrics Society. (2008) ;56: (10):1898–903. |
[283] | Marioni RE , Stewart MC , Murray GD , et al. Peripheral levels of fibrinogen, C-reactive protein, and plasma viscosity predict future cognitive decline in individuals without dementia. PsychosomMed. (2009) ;71: (8):901–6. |
[284] | Bruehl H , Wolf OT , Sweat V , Tirsi A , Richardson S , Convit A . Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. (2009) ;1280: :186–94. |
[285] | Bartels K , Grenz A , Eltzschig HK . Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci U S A. (2013) ;110: (46):18351–2. |
[286] | Meraz-Rios MA , Toral-Rios D , Franco-Bocanegra D , Villeda-Hernandez J , Campos-Pena V . Inflammatory process in Alzheimer’s Disease. Front Integr Neurosci. (2013) ;7: :59. |
[287] | Liguori I , Russo G , Curcio F , et al. Oxidative stress, aging, and diseases. Clin Interv Aging. (2018) ;13: :757–72. |
[288] | Hajjar I , Hayek SS , Goldstein FC , Martin G , Jones DP , Quyyumi A . Oxidative stress predicts cognitive decline with aging in healthy adults: An observational study. J Neuroinflammation. (2018) ;15: (1):17. |
[289] | Otaegui-Arrazola A , Amiano P , Elbusto A , Urdaneta E , Martinez-Lage P . Diet, cognition, and Alzheimer’s disease: Food for thought. Eur J Nutr. (2014) ;53: (1):1–23. |
[290] | Procaccini C , Santopaolo M , Faicchia D , et al. Role of metabolism in neurodegenerative disorders. Metabolism. (2016) ;65: (9):1376–90. |
[291] | Morgen K , Frolich L . The metabolism hypothesis of Alzheimer’s disease: From the concept of central insulin resistance and associated consequences to insulin therapy. J Neural Transm (Vienna). (2015) ;122: (4):499–504. |
[292] | Sieber BA , Landis S , Koroshetz W , et al. Prioritized research recommendations from the National Institute of Neurological Disorders and Stroke Parkinson’s Disease conference. Ann Neurol. (2014) ;76: (4):469–72. |
[293] | Corriveau RA , Koroshetz WJ , Gladman JT , et al. Alzheimer’s Disease-Related Dementias Summit National research priorities. Neurology. (2017) ;89: (23):2381–91. |
[294] | Singh-Manoux A , Dugravot A , Shipley M , et al. Obesity trajectories and risk of dementia: 28 years of follow-up in the Whitehall II Study. Alzheimers Dement. (2018) ;14: (2):178–86. |
[295] | Xu WL , Atti AR , Gatz M , Pedersen NL , Johansson B , Fratiglioni L . Midlife overweight and obesity increase late-life dementia risk: A population-based twin study. Neurology. (2011) ;76: (18):1568–74. |
[296] | Ronan L , Alexander-Bloch AF , Wagstyl K , et al. Obesity associated with increased brain age from midlife. Neurobiol Aging. (2016) ;47: :63–70. |
[297] | Dahl AK , Hassing LB . Obesity and cognitive aging. Epidemiol Rev. (2013) ;35: :22–32. |
[298] | Alford S , Patel D , Perakakis N , Mantzoros CS . Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes Rev. (2018) ;19: (2):269–80. |
[299] | Norton S , Matthews FE , Barnes DE , Yaffe K , Brayne C . Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. (2014) ;13: (8):788–94. |
[300] | Meng XF , Yu JT , Wang HF , et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: A systematic review and meta-analysis. J Alzheimers Dis. (2014) ;42: (4):1295–310. |
[301] | Exalto LG , Quesenberry CP , Barnes D , Kivipelto M , Biessels GJ , Whitmer RA . Midlife risk score for the prediction of dementia four decades later. Alzheimers Dement. (2014) ;10: (5):562–70. |
[302] | Anstey KJ , Cherbuin N , Budge M , Young J . Body mass index in midlife and late-life as a risk factor for dementia: A meta-analysis of prospective studies. Obes Rev. (2011) ;12: (5):e426–437. |
[303] | Tzourio C , Dufouil C , Ducimetiere P , Alperovitch A . Cognitive decline in individuals with high blood pressure: A longitudinal study in the elderly. EVA Study Group. Epidemiology of Vascular Aging. Neurology. (1999) ;53: (9):1948–52. |
[304] | Kuo H-K , Jones RN , Milberg WP , et al. Effect of blood pressure and diabetes mellitus on cognitive and physical functions in older adults: A longitudinal analysis of the advanced cognitive training for independent and vital elderly cohort. Journal of the American Geriatrics Society. (2005) ;53: (7):1154–61. |
[305] | Duron E , Hanon O . Hypertension, cognitive decline and dementia. Arch CardiovascDis. (2008) ;101: (3):181–9. |
[306] | Kennelly SP , Lawlor BA , Kenny RA . Blood pressure and the risk for dementia: A double edged sword. Ageing Res Rev. (2009) ;8: (2):61–70. |
[307] | Kennelly SP , Lawlor BA , Kenny RA . Blood pressure and dementia - a comprehensive review. Ther Adv Neurol Disord. (2009) ;2: (4):241–60. |
[308] | Wu C , Zhou D , Wen C , Zhang L , Como P , Qiao Y . Relationship between blood pressure and Alzheimer’s disease in Linxian County, China. Life Sci. (2003) ;72: (10):1125–33. |
[309] | Launer LJ , Ross GW , Petrovitch H , et al. Midlife blood pressure and dementia: The Honolulu-Asia aging study. NeurobiolAging. (2000) ;21: (1):49–55. |
[310] | Elias MF , Elias PK , Sullivan LM , Wolf PA , D’Agostino RB . Lower cognitive function in the presence of obesity and hypertension: The Framingham heart study. IntJ ObesRelat Metab Disord. (2003) ;27: (2):260–8. |
[311] | Wolf PA , Beiser A , Elias MF , Au R , Vasan RS , Seshadri S . Relation of obesity to cognitive function: Importance of central obesity and synergistic influence of concomitant hypertension. The Framingham Heart Study. CurrAlzheimer Res. (2007) ;4: (2):111–6. |
[312] | McGregor G , Harvey J . Food for thought: Leptin regulation of hippocampal function and its role in Alzheimer’s disease. Neuropharmacology. (2018) ;136: (Pt B):298–306. |
[313] | Fleckenstein J , Matura S , Engeroff T , et al. SMART: Physical activity and cerebral metabolism in older people: Study protocol for a randomised controlled trial. Trials. (2015) ;16: :155. |
[314] | Scarmeas N , Luchsinger JA , Schupf N , et al. Physical activity, diet, and risk of Alzheimer disease. Jama. (2009) ;302: (6):627–37. |
[315] | Veronese N , Facchini S , Stubbs B , et al. Weight loss is associated with improvements in cognitive function among overweight and obese people: A systematic review and meta-analysis. Neurosci Biobehav Rev. (2017) ;72: :87–94. |
[316] | Heni M , Kullmann S , Preissl H , Fritsche A , Haring HU . Impaired insulin action in the human brain: Causes and metabolic consequences. Nat Rev Endocrinol. (2015) ;11: (12):701–11 . |
[317] | Sartorius T , Heni M , Tschritter O , et al. Leptin affects insulin action in astrocytes and impairs insulin-mediated physical activity. Cell Physiol Biochem. (2012) ;30: (1):238–46. |
[318] | Cai H , Cong WN , Ji S , Rothman S , Maudsley S , Martin B . Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res. (2012) ;9: (1):5–17. |
[319] | Kandola A , Hendrikse J , Lucassen PJ , Yucel M . Aerobic Exercise as a Tool to Improve Hippocampal Plasticity and Function in Humans: Practical Implications for Mental Health Treatment. Frontiers in Human Neuroscience. (2016) ;10: :373. |
[320] | Vaynman S , Ying Z , Wu A , Gomez-Pinilla F . Coupling energy metabolism with a mechanism to support brain-derived neurotrophic factor-mediated synaptic plasticity. Neuroscience. (2006) ;139: (4):1221–34. |
[321] | Lin TW , Tsai SF , Kuo YM . Physical Exercise Enhances Neuroplasticity and Delays Alzheimer’s Disease. Brain Plast. (2018) ;4: (1):95–110. |
[322] | Ferreira AC , Sousa N , Bessa JM , Sousa JC , Marques F . Metabolism and adult neurogenesis: Towards an understanding of the role of lipocalin-2 and iron-related oxidative stress. Neurosci Biobehav Rev. (2018) ;95: :73–84. |
[323] | Beckervordersandforth R . Mitochondrial Metabolism-Mediated Regulation of Adult Neurogenesis. Brain Plast. (2017) ;3: (1):73–87. |
[324] | Knobloch M , Jessberger S . Metabolism and neurogenesis. Curr Opin Neurobiol. (2017) ;42: :45–52. |
[325] | Vilar M , Mira H . Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front Neurosci. (2016) ;10: :26. |
[326] | Fan LW , Pang Y . Dysregulation of neurogenesis by neuroinflammation: Key differences in neurodevelopmental and neurological disorders. Neural Regen Res. (2017) ;12: (3):366–71. |
[327] | Fuster-Matanzo A , Llorens-Martin M , Hernandez F , Avila J . Role of neuroinflammation in adult neurogenesis and Alzheimer disease: Therapeutic approaches. Mediators Inflamm. (2013) ;2013: :260925. |
[328] | Pereira AC , Huddleston DE , Brickman AM , et al. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. ProcNatlAcadSci US A. (2007) ;104: (13):5638–43. |
[329] | Mastrorilli V , Scopa C , Saraulli D , et al. Physical exercise rescues defective neural stem cells and neurogenesis in the adult subventricular zone of Btg1 knockout mice. Brain Struct Funct. (2017) ;222: (6):2855–76. |
[330] | Apple DM , Fonseca RS , Kokovay E . The role of adult neurogenesis in psychiatric and cognitive disorders. Brain Res. (2017) ;1655: :270–6. |
[331] | Lamm O , Ganz J , Melamed E , Offen D . Harnessing neurogenesis for the possible treatment of Parkinson’s disease. J Comp Neurol. (2014) ;522: (12):2817–30. |
[332] | Bedard C , Wallman MJ , Pourcher E , Parent A , Parent M . Intense dopamine innervation of the subventricular zone in Huntington’s disease. Neuroreport. (2010) ;21: (17):1074–9. |
[333] | Lennington JB , Pope S , Goodheart AE , et al. Midbrain dopamine neurons associated with reward processing innervate the neurogenic subventricular zone. J Neurosci. (2011) ;31: (37):13078–13087. |
[334] | Kleinridders A , Cai W , Cappellucci L , et al. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc Natl Acad Sci U S A. (2015) ;112: (11):3463–8. |
[335] | Yin L , Xu X , Chen G , et al. Inflammation and decreased functional connectivity in a widely-distributed network in depression: Centralized effects in the ventral medial prefrontal cortex. Brain Behav Immun. (2019) . |
[336] | Felger JC , Li Z , Haroon E , et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. (2016) ;21: (10):1358–65. |
[337] | Rendeiro C , Rhodes JS , Spencer JP . The mechanisms of action of flavonoids in the brain: Direct versus indirect effects. Neurochem Int. (2015) ;89: :126–39. |
[338] | Baumgart M , Snyder HM , Carrillo MC , Fazio S , Kim H , Johns H . Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement. (2015) ;11: (6):718–26. |
[339] | Schneider N , Yvon C . A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. (2013) ;17: (3):252–257. |
[340] | Akinyemi RO , Mukaetova-Ladinska EB , Attems J , Ihara M , Kalaria RN . Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr Alzheimer Res. (2013) ;10: (6):642–53. |
[341] | Booth V , Hood V , Kearney F . Interventions incorporating physical and cognitive elements to reduce falls risk in cognitively impaired older adults: A systematic review. JBI Database System Rev Implement Rep. (2016) ;14: (5):110–35. |
[342] | Eggenberger P , Schumacher V , Angst M , Theill N , de Bruin ED . Does multicomponent physical exercise with simultaneous cognitive training boost cognitive performance in older adults? A 6-month randomized controlled trial with a 1-year follow-up. Clin Interv Aging. (2015) ;10: :1335–49. |
[343] | Eggenberger P , Theill N , Holenstein S , Schumacher V , de Bruin ED . Multicomponent physical exercise with simultaneous cognitive training to enhance dual-task walking of older adults: A secondary analysis of a 6-month randomized controlled trial with 1-year follow-up. Clin Interv Aging. (2015) ;10: :1711–32. |
[344] | Cesari M , Demougeot L , Boccalon H , Guyonnet S , Vellas B , Andrieu S . The Multidomain Intervention to preveNt disability in ElDers (MINDED) project: Rationale and study design of a pilot study. Contemp Clin Trials. (2014) ;38: (1):145–54. |
[345] | Cerami C , Dodich A , Iannaccone S , et al. A biomarker study in long-lasting amnestic mild cognitive impairment. Alzheimers Res Ther. (2018) ;10: (1):42. |
[346] | Demetrius LA , Driver J . Alzheimer’s as a metabolic disease. Biogerontology. (2013) ;14: (6):641–9. |
[347] | Letra L , Santana I , Seica R . Obesity as a risk factor for Alzheimer’s disease: The role of adipocytokines. Metab Brain Dis. (2014) ;29: (3):563–8. |
[348] | Fadel JR , Jolivalt CG , Reagan LP . Food for thought: The role of appetitive peptides in age-related cognitive decline. Ageing Res Rev. (2013) ;12: (3):764–76. |
[349] | Arnold SE , Arvanitakis Z , Macauley-Rambach SL , et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat Rev Neurol. (2018) ;14: (3):168–81. |
[350] | Wijesekara N , Goncalves RA , De Felice FG , Fraser PE . Impaired peripheral glucose homeostasis and Alzheimer’s disease. Neuropharmacology. (2017) . |
[351] | Mencken HL . Prejudices: Second Series. London: Jonathan Cape; (1921) . |
[352] | Tim Minchin and the Power of Language [press release]. University of Western Australia2013. |
[353] | Valeri L , Vanderweele TJ . Mediation analysis allowing for exposure-mediator interactions and causal interpretation: Theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. (2013) ;18: (2):137–50. |
[354] | Vanderweele TJ , Vansteelandt S , Robins JM . Effect decomposition in the presence of an exposure-induced mediator-outcome confounder. Epidemiology. (2014) ;25: (2):300–6. |
[355] | Collins LM , Murphy SA , Strecher V . The multiphase optimization strategy (MOST) and the sequential multiple assignment randomized trial (SMART): New methods for more potent eHealth interventions. American journal of preventive medicine. (2007) ;32: (5 Suppl):S112–118. |
[356] | Collins LM , Murphy SA , Nair VN , Strecher VJ . A strategy for optimizing and evaluating behavioral interventions. Ann Behav Med. (2005) ;30: (1):65–73. |
[357] | Hopkins ME , Davis FC , Vantieghem MR , Whalen PJ , Bucci DJ . Differential effects of acute and regular physical exercise on cognition and affect. Neuroscience. (2012) ;215: :59–68. |
[358] | Ricci G , Pirillo I , Tomassoni D , Sirignano A , Grappasonni I . Metabolic syndrome, hypertension, and nervous system injury: Epidemiological correlates. Clin Exp Hypertens. (2017) ;39: (1):8–16. |
[359] | Best JR , Rosano C , Aizenstein HJ , et al. Long-term changes in time spent walking and subsequent cognitive and structural brain changes in older adults. Neurobiol Aging. (2017) ;57: :153–61. |
[360] | Jayaraman A , Pike CJ . Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr Diab Rep. (2014) ;14: (4):476. |
[361] | Hayes AF , Rockwood NJ . Regression-based statistical mediation and moderation analysis in clinical research: Observations, recommendations, and implementation. Behav Res Ther. (2017) ;98: :39–57. |
[362] | Preacher KJ , Hayes AF . Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods. (2008) ;40: (3):879–91. |
[363] | O’Rourke HP , MacKinnon DP . Reasons for Testing Mediation in the Absence of an Intervention Effect: A Research Imperative in Prevention and Intervention Research. J Stud Alcohol Drugs. (2018) ;79: (2):171–81. |
[364] | VanderWeele TJ , Vansteelandt S . Mediation Analysis with Multiple Mediators. Epidemiol Methods. (2014) ;2: (1):95–115. |
[365] | Gates N , Fiatarone Singh MA , Sachdev PS , Valenzuela M . The effect of exercise training on cognitive function in older adults with mild cognitive impairment: A meta-analysis of randomized controlled trials. The American Journal of Geriatric Psychiatry: Official Journal of the American Association for Geriatric Psychiatry. (2013) ;21: (11):1086–97. |
[366] | Zheng G , Xia R , Zhou W , Tao J , Chen L . Aerobic exercise ameliorates cognitive function in older adults with mild cognitive impairment: A systematic review and meta-analysis of randomised controlled trials. Br J Sports Med. (2016) . |
[367] | McDonnell MN , Smith AE , Mackintosh SF . Aerobic exercise to improve cognitive function in adults with neurological disorders: A systematic review. Arch Phys Med Rehabil. (2011) ;92: (7):1044–52. |
[368] | Nikseresht M , Agha-Alinejad H , Azarbayjani MA , Ebrahim K . Effects of nonlinear resistance and aerobic interval training on cytokines and insulin resistance in sedentary men who are obese. J Strength Cond Res. (2014) ;28: (9):2560–8. |
[369] | Bird SR , Hawley JA . Update on the effects of physical activity on insulin sensitivity in humans. BMJ Open Sport Exerc Med. (2016) ;2: (1):e000143. |
[370] | VanderWeele TJ , Tchetgen Tchetgen EJ . Mediation analysis with time varying exposures and mediators. J R Stat Soc Series B Stat Methodol. (2017) ;79: (3):917–38. |
[371] | Angevaren M , Aufdemkampe G , Verhaar HJJ , Aleman A , Vanhees L . Physical activity and enhanced fitness to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst Rev. 2008: (3):CD005381. |
[372] | Lamb SE , Sheehan B , Atherton N , et al. Dementia And Physical Activity (DAPA) trial of moderate to high intensity exercise training for people with dementia: Randomised controlled trial. BMJ. (2018) ;361: :k1675. |
[373] | Picano E , Bruno RM , Ferrari GF , Bonuccelli U . Cognitive impairment and cardiovascular disease: So near, so far. Int J Cardiol. (2014) ;175: (1):21–9. |
[374] | Driscoll I , Gaussoin SA , Wassertheil-Smoller S , et al. Obesity and Structural Brain Integrity in Older Women: The Women’s Health Initiative Magnetic Resonance Imaging Study. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2016) . |
[375] | Pruzin JJ , Nelson PT , Abner EL , Arvanitakis Z . Invited Review: Relationship of type 2 diabetes to human brain pathology. Neuropathol Appl Neurobiol. (2018) . |
[376] | Dichgans M , Leys D . Vascular Cognitive Impairment. Circ Res. (2017) ;120: (3):573–91. |
[377] | Wolters FJ , Zonneveld HI , Hofman A , et al. Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation. (2017) . |
[378] | Luchsinger JA , Ma Y , Christophi CA , et al. Metformin, Lifestyle Intervention, and Cognition in the Diabetes Prevention Program Outcomes Study. Diabetes Care. (2017) ;40: (7):958–65. |
[379] | Middleton LE , Yaffe K . Targets for the prevention of dementia. J Alzheimers Dis. (2010) ;20: (3):915–24. |
[380] | Middleton LE , Yaffe K . Promising strategies for the prevention of dementia. Arch Neurol. (2009) ;66: (10):1210–5. |
[381] | Tadic M , Cuspidi C , Hering D . Hypertension and cognitive dysfunction in elderly: Blood pressure management for this global burden. BMC Cardiovasc Disord. (2016) ;16: (1):208. |
[382] | de la Torre JC . Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer’s disease. J Alzheimers Dis. (2012) ;32: (3):553–67. |
[383] | Miquel S , Champ C , Day J , et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res Rev. (2018) ;42: :40–55. |
[384] | Morbelli S , Perneczky R , Drzezga A , et al. Metabolic networks underlying cognitive reserve in prodromal Alzheimer disease: A European Alzheimer disease consortium project. J Nucl Med. (2013) ;54: (6):894–902. |
[385] | Prehn K , Jumpertz von Schwartzenberg R , Mai K , et al. Caloric Restriction in Older Adults-Differential Effects of Weight Loss and Reduced Weight on Brain Structure and Function. Cereb Cortex. (2017) ;27: (3):1765–78. |
[386] | Alvarez JA , Emory E . Executive function and the frontal lobes: A meta-analytic review. Neuropsychol Rev. (2006) ;16: (1):17–42. |
[387] | Ardila A , Bernal B , Rosselli M . Executive Functions Brain System: An Activation Likelihood Estimation Meta-analytic Study. Arch Clin Neuropsychol. (2018) ;33: (4):379–405. |
[388] | Fjell AM , Westlye LT , Grydeland H , et al. Accelerating cortical thinning: Unique to dementia or universal in aging? Cerebral cortex. (2014) ;24: (4):919–34. |
[389] | Tamnes CK , Walhovd KB , Dale AM , et al. Brain development and aging: Overlapping and unique patterns of change. Neuroimage. (2013) ;68: :63–74. |
[390] | Carmichael O , Schwarz C , Drucker D , et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol. (2010) ;67: (11):1370–8. |
[391] | Fjell AM , Westlye LT , Espeseth T , et al. Cortical gray matter atrophy in healthy aging cannot be explained by undetected incipient cognitive disorders: A comment on Burgmans et al. (2009). Neuropsychology. (2010) ;24: (2):258–263; discussion 264–266. |
[392] | Fjell AM , Walhovd KB , Fennema-Notestine C , et al. One-year brain atrophy evident in healthy aging. J Neurosci. (2009) ;29: (48):15223–31. |
[393] | Soderlund H , Nyberg L , Adolfsson R , Nilsson LG , Launer LJ . High prevalence of white matter hyperintensities in normal aging: Relation to blood pressure and cognition. Cortex. (2003) ;39: (4-5):1093–105. |
[394] | Jellinger KA . The pathology of ischemic-vascular dementia: An update. JNeurolSci. (2002) ;203-204: :153–7. |
[395] | Jellinger KA . Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front Aging Neurosci. (2013) ;5: :17. |
[396] | Daianu M , Jahanshad N , Nir TM , et al. Breakdown of brain connectivity between normal aging and Alzheimer’s disease: A structural k-core network analysis. Brain Connect. (2013) ;3: (4):407–22. |
[397] | Lockhart SN , DeCarli C . Structural imaging measures of brain aging. Neuropsychol Rev. (2014) ;24: (3):271–89. |
[398] | Menon V , Uddin LQ . Saliency, switching, attention and control: A network model of insula function. Brain Struct Funct. (2010) ;214: (5-6):655–67. |
[399] | Raichle ME . The brain’s default mode network. Annu Rev Neurosci. (2015) ;38: :433–47. |
[400] | Fox MD , Zhang D , Snyder AZ , Raichle ME . The global signal and observed anticorrelated resting state brain networks. J Neurophysiol. (2009) ;101: (6):3270–83. |
[401] | Ramani R . Connectivity. Curr Opin Anaesthesiol. (2015) ;28: (5):498–504. |
[402] | Goulden N , Khusnulina A , Davis NJ , et al. The salience network is responsible for switching between the default mode network and the central executive network: Replication from DCM. Neuroimage. (2014) ;99: :180–90. |
[403] | Hasenkamp W , Wilson-Mendenhall CD , Duncan E , Barsalou LW . Mind wandering and attention during focused meditation: A fine-grained temporal analysis of fluctuating cognitive states. Neuroimage. (2012) ;59: (1):750–60. |
[404] | Weng TB , Pierce GL , Darling WG , Falk D , Magnotta VA , Voss MW . The Acute Effects of Aerobic Exercise on the Functional Connectivity of Human Brain Networks. Brain Plast. (2017) ;2: (2):171–90. |
[405] | Garcia-Agundez A , Folkerts AK , Konrad R , et al. Recent advances in rehabilitation for Parkinson’s Disease with Exergames: A Systematic Review. J Neuroeng Rehabil. (2019) ;16: (1):17. |
[406] | Uc EY , Doerschug KC , Magnotta V , et al. Phase I/II randomized trial of aerobic exercise in Parkinson disease in a community setting. Neurology. (2014) ;83: (5):413–25. |
[407] | Metcalfe AW , MacIntosh BJ , Scavone A , Ou X , Korczak D , Goldstein BI . Effects of acute aerobic exercise on neural correlates of attention and inhibition in adolescents with bipolar disorder. Transl Psychiatry. (2016) ;6: :e814. |
[408] | Hoffmann M . The human frontal lobes and frontal network systems: An evolutionary, clinical, and treatment perspective. ISRN Neurol. (2013) ;2013: :892459. |
[409] | Ramos BP , Arnsten AF . Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol Ther. (2007) ;113: (3):523–36. |
[410] | Clewett DV , Lee TH , Greening S , Ponzio A , Margalit E , Mather M . Neuromelanin marks the spot: Identifying a locus coeruleus biomarker of cognitive reserve in healthy aging. Neurobiol Aging. (2016) ;37: :117–26. |
[411] | Robertson IH . A noradrenergic theory of cognitive reserve: Implications for Alzheimer’s disease. Neurobiol Aging. (2013) ;34: (1):298–308. |
[412] | Berse T , Rolfes K , Barenberg J , et al. Acute physical exercise improves shifting in adolescents at school: Evidence for a dopaminergic contribution. Front Behav Neurosci. (2015) ;9: :196. |
[413] | Lee YY , Wu CY , Teng CH , Hsu WC , Chang KC , Chen P . Evolving methods to combine cognitive and physical training for individuals with mild cognitive impairment: Study protocol for a randomized controlled study. Trials. (2016) ;17: (1):526. |
[414] | Khalsa DS , Perry G . The Four Pillars of Alzheimer’s Prevention. Cerebrum. (2017) ;2017. |
[415] | Kivipelto M , Mangialasche F , Ngandu T , World Wide Fingers N. World Wide Fingers will advance dementia prevention. Lancet Neurol. (2018) ;17: (1):27. |
[416] | Barnes DE , Santos-Modesitt W , Poelke G , et al. The Mental Activity and eXercise (MAX) Trial: A Randomized Controlled Trial to Enhance Cognitive Function in Older Adults. JAMA Intern Med. (2013) :1–8. |
[417] | Erickson KI , Creswell JD , Verstynen TD , Gianaros PJ . Health Neuroscience: Defining a New Field. Curr Dir Psychol Sci. (2014) ;23: (6):446–53. |
[418] | Mullers P , Taubert M , Muller NG . Physical Exercise as Personalized Medicine for Dementia Prevention? Front Physiol. (2019) ;10: :672. |
[419] | Lo RY , Jagust WJ , Alzheimer’s Disease Neuroimaging I. Effect of cognitive reserve markers on Alzheimer pathologic progression. Alzheimer Dis Assoc Disord. (2013) ;27: (4):343–50. |
[420] | Pearl J . The causal mediation formula–a guide to the assessment of pathways and mechanisms. Prev Sci. (2012) ;13: (4):426–36. |
[421] | Nascimento CM , Pereira JR , de Andrade LP , et al. Physical exercise in MCI elderly promotes reduction of pro-inflammatory cytokines and improvements on cognition and BDNF peripheral levels. Curr Alzheimer Res. (2014) ;11: (8):799–805. |
[422] | Melah KE , Lu SY , Hoscheidt SM , et al. Cerebrospinal Fluid Markers of Alzheimer’s Disease Pathology and Microglial Activation are Associated with Altered White Matter Microstructure in Asymptomatic Adults at Risk for Alzheimer’s Disease. J Alzheimers Dis. (2016) ;50: (3):873–86. |
[423] | Dai Y , Kamal MA . Fighting Alzheimer’s disease and type 2 diabetes: Pathological links and treatment strategies. CNS Neurol Disord Drug Targets. (2014) ;13: (2):271–82. |
[424] | Tucsek Z , Toth P , Sosnowska D , et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2014) ;69: (10):1212–26. |
[425] | Srodulski S , Sharma S , Bachstetter AB , et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener. (2014) ;9: :30. |
[426] | Palmqvist S , Minthon L , Wattmo C , Londos E , Hansson O . A Quick Test of cognitive speed is sensitive in detecting early treatment response in Alzheimer’s disease. Alzheimers Res Ther. (2010) ;2: (5):29. |
[427] | Wattmo C , Wallin AK , Minthon L . Progression of mild Alzheimer’s disease: Knowledge and prediction models required for future treatment strategies. Alzheimers Res Ther. (2013) ;5: (5):44. |
[428] | Wall J , Xie H , Wang X . An Exploration Into Short-Interval Maintenance of Adult Hemispheric Cortical Thickness at an Individual Brain Level. J Exp Neurosci. (2017) ;11: :1179069517733453. |
[429] | Storsve AB , Fjell AM , Tamnes CK , et al. Differential longitudinal changes in cortical thickness, surface area and volume across the adult life span: Regions of accelerating and decelerating change. J Neurosci. (2014) ;34: (25):8488–98. |
[430] | Macdonald KE , Bartlett JW , Leung KK , Ourselin S , Barnes J , investigators A . The value of hippocampal and temporal horn volumes and rates of change in predicting future conversion to AD. Alzheimer Dis Assoc Disord. (2013) ;27: (2):168–73. |
[431] | Leung KK , Bartlett JW , Barnes J , et al. Cerebral atrophy in mild cognitive impairment and Alzheimer disease: Rates and acceleration. Neurology. (2013) ;80: (7):648–54. |
[432] | Kalin AM , Park MT , Chakravarty MM , et al. Subcortical Shape Changes, Hippocampal Atrophy and Cortical Thinning in Future Alzheimer’s Disease Patients. Front Aging Neurosci. (2017) ;9: :38. |
[433] | Matsuda H . Voxel-based Morphometry of Brain MRI in Normal Aging and Alzheimer’s Disease. Aging Dis. (2013) ;4: (1):29–37. |
[434] | Barnes J , Carmichael OT , Leung KK , et al. Vascular and Alzheimer’s disease markers independently predict brain atrophy rate in Alzheimer’s Disease Neuroimaging Initiative controls. Neurobiol Aging. (2013) ;34: (8):1996–2002. |
[435] | Xue W , Bowman FD , Kang J . A Bayesian Spatial Model to Predict Disease Status Using Imaging Data From Various Modalities. Front Neurosci. (2018) ;12: :184. |
[436] | Zhang L , Wang M , Sterling NW , et al. Cortical Thinning and Cognitive Impairment in Parkinson’s Disease without Dementia. IEEE/ACM Trans Comput Biol Bioinform. (2018) ;15: (2):570–80. |
[437] | Patel RS , Bowman FD , Rilling JK . Determining hierarchical functional networks from auditory stimuli fMRI. Hum Brain Mapp. (2006) ;27: (5):462–70. |
[438] | Patel RS , Bowman FD , Rilling JK . A Bayesian approach to determining connectivity of the human brain. Hum Brain Mapp. (2006) ;27: (3):267–76. |
[439] | Chen S , Bowman FD , Mayberg HS . A Bayesian hierarchical framework for modeling brain connectivity for neuroimaging data. Biometrics. (2016) ;72: (2):596–605. |
[440] | Xue W , Bowman FD , Pileggi AV , Mayer AR . A multimodal approach for determining brain networks by jointly modeling functional and structural connectivity. Front Comput Neurosci. (2015) ;9: :22. |
[441] | Derado G , Bowman FD , Zhang L , Alzheimer’s Disease Neuroimaging Initiative I. Predicting brain activity using a Bayesian spatial model. Stat Methods Med Res. (2013) ;22: (4):382–97. |
[442] | Reed BR , Mungas D , Farias ST , et al. Measuring cognitive reserve based on the decomposition of episodic memory variance. Brain. (2010) ;133: (Pt 8):2196–209. |
[443] | Zahodne LB , Manly JJ , Brickman AM , et al. Is residual memory variance a valid method for quantifying cognitive reserve? A longitudinal application. Neuropsychologia. (2015) ;77: :260–6. |
[444] | Zahodne LB , Manly JJ , Brickman AM , Siedlecki KL , Decarli C , Stern Y . Quantifying cognitive reserve in older adults by decomposing episodic memory variance: Replication and extension. J Int Neuropsychol Soc. (2013) ;19: (8):854–62. |
[445] | Petkus AJ , Resnick SM , Rapp SR , et al. General and domain-specific cognitive reserve, mild cognitive impairment, and dementia risk in older women. Alzheimers Dement (N Y). (2019) ;5: :118–28. |
[446] | Wee CY , Yap PT , Denny K , et al. Resting-state multi-spectrum functional connectivity networks for identification of MCI patients. PLoS One. (2012) ;7: (5):e37828. |
[447] | Wee CY , Yap PT , Zhang D , et al. Identification of MCI individuals using structural and functional connectivity networks. Neuroimage. (2012) ;59: (3):2045–56. |
[448] | Badhwar A , Tam A , Dansereau C , Orban P , Hoffstaedter F , Bellec P . Resting-state network dysfunction in Alzheimer’s disease: A systematic review and meta-analysis. Alzheimers Dement (Amst). (2017) ;8: :73–85. |

